

Topamax dosages: 200 mg, 100 mg
Topamax packs: 30 pills, 60 pills, 90 pills, 120 pills, 180 pills, 270 pills
Discount topamax 200 mg overnight delivery
An toddler in one other family had hyperinsulinism that was readily managed with diazoxide and hydrochlorthiazide [11] medications hyperkalemia topamax 200 mg order. Analysis of the free fatty acids of the plasma or organic acid analysis of the urine may reveal medium-chain 3-hydroxy fatty acids treatment vertigo 200 mg topamax, even when the patient is metabolically well [12, 13]. However, this pattern may be seen in infants receiving mediumchain triglycerides. The focus of free carnitine within the plasma may be normal or low, and there could also be elevated quantities of carnitine esters within the urine. Adipic, suberic, and sebacic acids are discovered, as nicely as 3-hydroxydicarboxylic acids. In our patient, challenge with a load of medium-chain triglyceride led to elevated excretion of dicarboxylic acids and 3-hydroxydicarboxylic acids. In one patient [9], elevated C16 and C18 acylcarnitines have been found, and no hydroxyacylcarnitines. A novel homozygous mutation was found [15] in a family with extreme neonatal hypoglycemia and hyperinsulinemia. Treatment with glucagon and diazoxide has been employed in the extreme hyperinsulinemic hypoglycemic sufferers [15]. A hyperlink between fatty acid oxidation and the dysregulation of acid oxidation and the dysregulation of the secretion of insulin has been clarified by studies in hadh -/- mice [16]. Short-chain l-3-hydroxyacylCoA dehydrogenase deficiency in muscle: a brand new cause for recurrent myoglobinuria and encephalopathy. Purification and properties of enoyl-coenzyme A (CoA) hydratase/3hydroxyacyl-CoA dehydrogenase/3-ketoacyl-CoA thiolase trifunctional protein. Human short-chain L-3-hydroxyacyl-CoA dehydrogenase: cloning and characterization of the coding sequence. Consanguinity has been noticed and heterozygosity has been demonstrated by enzyme assay of the liver [9] and by mutational analysis [12]. The gene encodes a 34-kDa precursor protein that, with processing, yields a mature 31-kDa subunit. Deficiency of enzyme activity has been demonstrated in muscle [1], liver [9], and fibroblasts [12], as well as in mitochondria isolated from fibroblasts [11]. In some families, the faulty enzyme was not demonstrable in fibroblasts, but was present in muscle [1] or liver [9]. The human gene encodes a protein of 314 amino acids and is expressed within the liver, kidney, coronary heart, and muscle. Fulminant hepatic failure associated with mutations within the medium and short chain L-3-hydroxyacyl-CoA dehydrogenase gene. Short-chain hydroxyacylcoenzyme A dehydrogenase deficiency presenting as surprising infant demise: a family historical past. Mitochondrial shortchain L-3-hydroxyacyl-coenzyme A dehydrogenase deficiency: a new defect of fatty acid oxidation. Hyperinsulinism in short-chain L-3-hydroxyacyl-CoA dehydrogenase deficiency reveals the importance of beta-oxidation in insulin secretion. Familial hyperinsulinemic hypoglycemia with a mutation within the gene encoding short-chain 3-hydroxyacyl-CoA dehydrogenase. The cblD defect causes eith isolated or mixed deficiency of methylcobalamin and adenosylcobalamin synthesis. Mechanism of hyperinsulinism in short-chain 3-hydroxyacyl-CoA dehydrogenase deficiency entails activation of glutamate dehydrogenase. Of two affected siblings in the initial report of Gibson and colleagues in 2000 [1], the first manifested neurologic abnormalities following a probable ischemic/hypoxic occasion at three days of age. His sister, identified prenatally, had been utterly asymptomatic by the point of follow-up report [2], and a selection of different patients have been asymptomatic, notably those identified by neonatal screening [2�5]. The key to the metabolic abnormality was the excretion of 2-methylbutyrylglycine in the urine and an elevated degree of 2-methylbutyrylcarnitine within the blood. By 12 months of age, he was behind in visual, motor, and cognitive abilities and carried a diagnosis of athetoid cerebral palsy. In one other household [2, 3], the patient was a three-year-old product of a consanguineous mating who had hypotonia and impaired motor improvement. Among ethnic Hmongs, eight sufferers have been discovered on expanded new child screening to have 2-methylbutyrylglycinuria and a distinct mutation [4]. Except for delicate muscular hypotonia noticed at six months of age, all were asymptomatic. Of two different consanguineous patients, one had attention-deficient dysfunction, and one convulsive illness and developmental delay. It appears that patients with this disorder could additionally be asymptomatic, but neurologic disease could also be a feature. Interelations of the S and R pathways of isoleucine catabolism the gene incorporates eleven exons [7]; its open reading frame of 1. Mutational evaluation in the first affected person revealed a C778T substitution within the coding area which led to the substitution of a phenylalanine for leucine at amino acid 22 [1]. Mutational analysis revealed homozygosity for a G1228A transition within the second affected person and his mom [3]. Mutational analysis in the Hmong patients yielded a homozygous A1165G mutation, which additionally led to skipping of exon 10. Incubation of to 2 13C-isoleucine with L-carnitine in intact cultured fibroblasts led to accumulation of isotope in C5-acylcarnitine. In the second patient [3], the activity of 2-methylbutyrylCoA dehydrogenase in fibroblasts was 10 p.c of management. Defective exercise was also demonstrated by expressing the abnormal gene merchandise in E. Many of those reported have been discovered via programs of expanded new child screening. Elevated C5 acylcarnitine could also be documented by analysis of the plasma, in addition to of dried blood on filter paper. Chiral willpower of 2-methylbutyric acid indicated that 40�46 p.c was in the form of the R isomer in patients and in controls. Keto-enol tautomeric racemization following or enamine tautomerization throughout, transamination is the source of alloisoleucine (Chapter 19). Ethylhydracrylic acid excretion in increased quantity can also be observed in ketosis [12], in 3-oxothiolase deficiency [13], in 2-methyl-3-hydroxybutyrylCoA dehydrogenase deficiency [14], in propionic acidemia [15], and methylmalonic acidemia, all defects in steps of the S pathway. It can also be found in ethylmalonic encephalopathy, hydroxyisobutyric aciduria, and in Barth syndrome. Prenatal analysis was achieved [1] by evaluation of 2-methylbutyrylglycine in amniotic fluid (0. A protein intake of 1�4 g/kg together with carnitine of 71 mg/kg led to a traditional excretion of 2-methylbutyrylglycine [1]. Isolated 2-methylbutyrylglycinuria attributable to short/branched-chain acyl-CoA dehydrogenase deficiency: identification of a new enzyme defect, decision of its molecular foundation, and evidence for distinct acyl-CoA dehydrogenases in isoleucine and valine metabolism.
Syndromes
- Thyroid function
- Vomiting
- Joint destruction
- Gonorrhea
- Sudden belly or back pain that gets worse or is very severe
- Phenytoin
- Wheat (mostly in children)
Discount topamax 200 mg amex
When the mutation is understood medications over the counter buy topamax 200 mg fast delivery, molecular strategies may be used for the detection of carriers medicine 10 day 2 times a day chart generic 200 mg topamax with visa. Intrauterine prognosis of the affected fetus has been completed in sorts A and B illness by way of assay of the related enzyme in amniotic cells in tradition [58]. It could additionally be useful to affirm the assay by assessment of 35S accumulation, because some heterozygotes have very low levels of enzyme in the usual assays. Chorionic villus materials has ample enzyme exercise and is available for the prenatal analysis of each of the forms of Sanfilippo disease [58�61]. Some different deletions have been reported, but most mutations have been missense [15, 16, 63�66]. Among Australian, Dutch, and German patients, the most common mutation was R245H [17, 64]. Other frequent mutations are Q380R, S66W, and 1080delC, all with the extreme phenotype. Among Polish sufferers, it was R74C [65], S66W in Sardinians [65], and 1091delC in Spaniards [66]. The gene for N-acetylglucosamine 6-sulfatase, on chromosome 12q14, the positioning of the defect in Sanfilippo sort D, has been cloned and encodes a protein of fifty two amino acids. The first mutation to be present in a consanguineous Pakistani household was a homozygous single base pair deletion c. Enzyme replacement remedy has been tried utilizing References 587 partially purified enzyme, leukocytes, and cultured fibroblasts [73], after which there may have been some changes in urinary mucopolysaccharides, however medical profit has not been evident except in gentle tissues [74]. Outcome measures have been studied in a seek for helpful displays for evaluating medical trials [76]. The Mullen and Leiter scales of cognitive development and the parental report of adaptive habits were judged best. Mental retardation related to acid mucopolysacchariduria (heparitin sulfate type). Biochemical heterogeneity of the Sanfilippo syndrome: preliminary characterization of two poor elements. Sanfilippo syndrome: profound deficiency of alphaacetylglucosaminidase activity in organs and skin fibroblasts from sort B patients. Sanfilippo syndrome kind C: deficiency of acetyl-CoA: -glucosaminide N-acetyltransferase in skin fibroblasts. Sanfilippo disease sort D: deficiency of N-acetylglucosamine 6-sulfate sulfatase required for heparan sulfate degradation. Cloning of the sulphamidase gene and identification of mutations in Sanfilippo A syndrome. Genetic heterogeneity and medical variability in the Sanfilippo syndrome (types A, B and C). A recessively inherited lethal disease in a Caribbean isolate � a sulfamidase deficiency. Intrafamilial variability in Hurler syndrome and Sanfilippo syndrome sort A: implications for analysis of new therapies. The Sanfilippo syndrome: a clinical and genetical examine of seventy five sufferers within the Netherlands (doctoral thesis). Sanfilippo A syndrome: sulfamidase deficiency in cultured pores and skin fibroblasts and liver. Purification and partial characterization of a-N-acetylglucosaminidase from human liver. Biosynthesis and maturation of a-N-acetylglucosaminidase in regular and Sanfilippo B fibroblasts. New biochemical subtype of the Sanfilippo syndrome: characterization of the storage materials in cultured fibroblasts of Sanfilippo C patients. Sanfilippo illness sort D; deficiency of N-acetylglucosamine-6-sulfatase required for heparan sulfate degradation. Sanfilippo illness type B: presence of fabric cross reacting with antibodies in opposition to a-N-acetylglucosaminidase. Intralysosomal formation and metabolic destiny of N-acetylglucosamine 6-sulfate from keratan sulfate. Clinical variability in Sanfilippo B illness: a report of six sufferers in two related sibships. First trimester prenatal analysis of metabolic illnesses: a survey of countries from the European Community. Sanfilippo syndrome type D: a spectrophotometric assay with prenatal diagnostic potential. Identification of molecular defects in Italian Sanfilippo A sufferers together with thirteen novel mutations. Mutation 1091delC is very prevalent in Spanish Sanfilippo syndrome sort A patients. Sanfilippo syndrome kind D: identification of the primary mutation in the N-acetylglucosamine-6sulphatase gene. Neuropsychological outcomes of several storage illnesses with and with out bone marrow transplantation. The excretion of keratan sulfate in the urine is the defining biochemical characteristic of patients with this disease, and keratan sulfate has been documented to accumulate in tissues [3�5]. Keratan sulfate consists of alternating galactose and N-acetylglucosamine residues; each could additionally be sulfated. This enzyme additionally catalyzes the elimination of sulfate moieties from N-acetylgalactosamine6-sulfate residues which may be present in chondroitin-6-sulfate (C6S), and this results in excretion in excess of chondroitin sulfate in Morquio syndrome. The enzyme also hydrolyzes the sulfate from the N-acetylgalactosamine-6-sulfate moieties that occur in chondroitin-6-sulfate. Clinical abnormalities 591 the gene for the galactose-6-sulfatase deficient in kind A Morquio illness has been cloned [13] and mapped to chromosome 16q 24. The gene for the -galactosidase faulty in sort B has been localized to chromosome three p21-cen [20]. The most attribute features of this syndrome are skeletal deformities and shortness of stature, which is especially short-trunked, although the long bones are also involved. There can additionally be a pronounced genu valgum, and sufferers typically have a semi-crouching stance. Prominence of the lower ribs may first bring the affected person in for medical session at 12 to 18 months of age. Illustrated are the standard pectus carnatum, deformed arms and legs, and stunted stature. In the second or third yr, sufferers develop awkward gaits and impaired development as skeletal deformities begin to be evident. Diagnostic delay is the rule and the prognosis may not be made until as late as 15 years [25, 26]. These patients have fine corneal opacities, which are often visible only on slit lamp examination, however may trigger a hazy cloudiness of the cornea [3, 27]. Progressive sensorineural or blended deafness usually begins within the second decade and is present uniformly after 20 years [29].
Discount topamax 200 mg
In addition medications migraine headaches buy topamax 200 mg on line, a tri-functional enzyme catalyzes 3-hydroxyacyl dehydrogenation symptoms hepatitis c topamax 100 mg without a prescription, 2-enoyl-CoA hydration, and 3-oxoacylCoA thiolysis [7]. Even on this disease, however, no less than 5 rare mutations are known [8], and no much less than one widespread mutation T199C is present in sufferers identified by newborn screening. Acylcarnitine profiling should lead to the prognosis in every of those disorders [9] (Table 34. Acylcarnitine profiling has additionally been employed within the evaluation of postmortem bile to identify patients with disorders of fatty acid oxidation initially thought to have sudden infant dying syndrome [10]. During the lengthy fast, patients should be monitored intently so that symptomatic hypoglycemia is avoided. In abnormalities of fatty acid oxidation, fasting have to be long enough to exhaust shops of glycogen and require the mobilization of fats and its oxidation. The specific enzyme assays for particular issues are technically demanding and never usually out there. Experience with the diagnosis and administration of issues of fatty acid oxidation was summarized in 1999 for a collection of 107 sufferers with a spectrum of disorders [13]. The severity of those diseases is indicated by the reality that only 57 of the 107 sufferers had been alive at report. An extra forty seven siblings had died in infancy for a complete of ninety seven deaths, 30 % in the first week of life and 69 percent before one year. These knowledge symbolize the significance of new child screening in prevention, as a result of the avoidance of fasting would forestall many deaths. Seventy-three % had been judged to have hepatic scientific presentations, which included hypoketotic hypoglycemia, hepatomegaly, Reye syndrome, and microscopic hepatic steatosis. In addition, a beforehand unrecognized defect within the transport of long-chain fatty acids has been reported to trigger acute liver failure and hypoketotic hypoglycemia [14]. The clue to the analysis was low concentrations of C14-18 fatty acids and elevated carnitine, a really uncommon sample, in biopsied liver. Skeletal muscle involvement in fifty one p.c of patients included myalgia, myopathy, and rhabdomyolysis with myoglobinuria. Treatment of problems of fatty acid oxidation [15] may be considered underneath two headings: acute administration of acute metabolic imbalance, and continual preventive remedy. Insulin, along with glucose, could additionally be necessary to keep normoglycemia, and a central line or portacath could also be required. It has been controversial in long-chain fatty acid problems due to a theoretical threat that accumulation of long-chain acyl carnitines could also be arrhythmogenic, as present in experimental conditions [16]. Our patients are supplied with letters indicating the necessity for parenteral glucose each time intercurrent illness or vomiting preclude the enteral route. Overnight fasting is minimized by means of oral corn starch (1 g/kg hour; 8 g/tbsp). Restriction of long-chain dietary fat is generally prudent as is long-term oral carnitine. Medium-chain triglyceride supplementation is therapeutic in long-chain fatty acid defects. In vitro fibroblast research in a affected person with C6-C10-dicarboxylic aciduria: Evidence for a defect in general acyl-CoA dehydrogenase. Medium-chain acyl-CoA dehydrogenase deficiency in children with nonketotic hypoglycemia and low carnitine levels. Automated tandem mass spectrometry for mass newborn screening for problems in fatty acid, organic acid, and amino acid metabolism. On the biologic origin of C6-C10-dicarboxylic and C6-C10-1-hydroxy monocarboxylic acids in human and rat with acyl-CoA dehydrogenation deficiencies: in vitro studies on the - and -1-oxidation of medium-chain (C6-C12) fatty acids in human and rat liver. Analysis of fatty acid oxidation intermediates in cultured fibroblasts to detect mitochondrial oxidation issues. A defect within the transport of long-chain fatty acids related to acute liver failure. Prophylaxis of early ventricular fibrillation by inhibition of acylcarnitine accumulation. Human liver longchain 3-hydroxyacyl-coenzyme A dehydrogenase is a multifunctional membrane-bound �-oxidation enzyme of mitochondria. Inborn errors of metabolism diagnosed in sudden demise cases by acylcarnitine analysis of postmortem bile. Deficiency of carnitine is frequent in these disorders in which fatty acyl CoA compounds accumulate which then kind esters with carnitine and are preferentially excreted within the urine. Carnitine deficiency may be profound in natural acidemias, such as propionic acidemia, for a similar purpose. The transport of carnitine into fibroblasts is inhibited by long- and medium-chain acylcarnitines [3], and this can be an additional think about carnitine deficiency in issues of fatty acid oxidation. Primary carnitine deficiency resulting from an abnormality within the synthesis of carnitine from protein-bound lysine has not but been noticed. Resuscitation required intubation, assisted air flow, and parenteral glucose and saline. Biopsy of the liver exhibits microvesicular lipid [10], a finding, like the relaxation of this scientific image, that might result in a diagnosis of Reye syndrome. Clinical chemistry in the acute hypoketotic episode can additionally be according to Reye syndrome, with hyperammonemia and increased ranges of transaminases. She offered at three months with hypoglycemic coma precipitated by an intercurrent illness and prolonged fasting. This episode left her with severe brain damage mirrored in her vacant expression. She died shortly after the image was taken because of complications of a gastrostomy. His demise at thirteen years highlights the harmful nature of the illness and the importance of close follow up of carnitine status and professional administration. Examination of the urine could reveal no ketonuria [1, 3], and this could strongly counsel the prognosis. However, in the peak of the hypoglycemic illness, there may be misleading ketonuria in any dysfunction of fatty acid oxidation. Cardiomyopathy is the opposite classic method by which this disorder presents [3, 7, 10] and could also be anticipated in any affected person not given the good factor about diagnosis and therapy with carnitine. It was the commonest presenting complaint in Genetics and pathogenesis 273 15 patients [6] and current in 100 percent of 20 early reported sufferers. The affected person of Waber and colleagues [10] reported with progressive cardiomyopathy and cardiac failure efficiently treated with carnitine was subsequently proven to have the transporter defect. Onset may be with quickly progressive coronary heart failure [3] or a murmur, and cardiomegaly could also be discovered on routine bodily examination or examination on the time of hypoglycemia. Roentgenograms and echocardiography reveal cardiac enlargement and increased thickness of the left ventricular wall.
Order 100 mg topamax
Demonstration of a Sandhoff disease-associated autosomal 50-kb deletion by filed inversion gel electrophoresis symptoms 6 days post embryo transfer 100 mg topamax buy free shipping. Bone marrow transplantation prolongs life span and ameliorates neurologic manifestations in Sandhoff illness mice medicine remix order 200 mg topamax visa. Delayed symptom onset and elevated life expectancy in Sandhoffdisease mice treated with N-butyldeoxynojirimycin. Type 2: Acute neuronopathic: Early infantile onset, hypertonicity, seizures, trismus, retroflexion of the pinnacle; splenomegaly; hepatomegaly; fast neurologic deterioration and demise between one and 24 months. Type 3: Subacute neuronopathic: Splenomegaly, hepatomegaly; childhood onset of neurologic manifestations � ataxia, spastic paraparesis, seizures, ophthalmoplegia; dying in childhood or adulthood if untreated. He identified the pathognomonic cells, which are now known as Gaucher cells, in a 32-year-old woman with huge enlargement of the spleen. This phenotype, now referred to as kind 1, was recognized in the Nineteen Fifties to be widespread in Ashkenazi Jews [3]. The acute neuronopathic early childish, sort 2, disease was described in 1927 [4, 5]. The perinatal deadly form of the illness is now considered to be a distinct type of type 2. Actually, genetic heterogeneity is such that there are rising numbers of intermediate and overlapping forms. The separate designations are useful as a result of sort 1 is so frequent and naturally the neonatal lethal illness could be very completely different. The defective enzyme is a lysosomal acid -glucosidase, lively in catalyzing the release of glucose from a number of substrates in addition to glucosylceramide. There is an activator of the enzyme, saposin C, which has a low molecular weight [13]. The kind 1 illness provides an attention-grabbing therapeutic model as a result of enzyme substitute therapy has been fairly profitable [18]. Abdominal distention had been progressive and aossicated with weak point and failure to thrive. �-Glucosidase exercise of fibroblasts was 9 mmol/ mg per hour or 5 percent of normal. As many as 25 p.c of affected people may be asymptomatic or have splenomegaly found incidental to an examination well into grownup life, even into the eighth and ninth a long time [19�22]. Severely affected people with sort 1 illness could die within the first or second decade. It may be so massive as to intrude with the intake of meals into the abdomen or to trigger dispareunia. A giant infarction may produce the image of an acute abdomen, along with hyperuricemia. The stomach enlargement caused by the hepatomegaly is evident even with the affected person fully clothed. The massive liver is normally not associated with liver illness, but there could additionally be elevated transaminases, cirrhosis, esophageal varices, or hepatic failure [25�27]. Hepatic infarction could current as an acute stomach disaster with a Budd-Chiari syndrome. One patient we saw was left with a large palpable notch in the center of the hepatic define. It may be accompanied by leukopenia and anemia, the total picture of hypersplenism, and resolves with splenectomy [28]. Late hematologic dysfunction in splenectomized sufferers could outcome from replacement of normal marrow with Gaucher cells. In many patients, they take the form of acute crises of pain, tenderness, redness, and swelling, mimicking acute osteomyelitis or the thrombotic crises of sickle cell disease. There could additionally be fever, leukocytosis, and elevation of the erythrocyte sedimentation rate. Areas of focal destruction of bone, osteonecrosis, or avascular necrosis happen within the absence of acute crises, particularly in the space of the femoral head [31]. Compression fracture of vertebral bodies is a common complication [31, 33�35], and there could additionally be radicular or spinal cord compression or kyphoscoliotic deformity. In addition, areas of severe lack of bone density could alternate with areas of osteosclerosis and focal infarctions. In addition to the results of anemia, splenic enlargement, and continual illness, resting power expenditure is elevated about 44 p.c [39]. Roentgenogram revealed virtually completely opaque lung fields with air bronchograms. The nervous system is by definition spared in type 1 disease, but a number of secondary issues do have an effect on the nervous system in addition to the problems of vertebral illness. These embody cerebral fats emboli secondary to skeletal disease, neuropathy and hematomyelia secondary to bleeding [42]; a affected person was reported with lumbosacral cauda equina syndrome secondary to an intrathecal sacral cyst, apparently the outcomes of subdural hemorrhage [44]. A few patients with sort 1 Gaucher disease have been found to develop parkinsonian signs [45], with onset typically within the fourth decade and a restricted responsiveness to remedy. There has not been any association with genotype, but in several instances, there was a family historical past of parkinsonism [46]. In a examine of tissue from patients with parkinsonism, alterations in glucocerebrosidase were present in 12 out of fifty seven individuals [47]. Malignancies including a number of myeloma and leukemia and monoclonal gammopathies have been reported in Gaucher illness [48, 49]. Multifocal jerky movements are widespread within the muscular tissues and occur at relaxation or with movement. Careful examination of the eyes discloses deficits in saccadic velocities, progressive with time to paralysis of lateral gaze [63, 64]. A group of sufferers in whom paralysis of horizontal supranuclear gaze is the main neurologic sign have been considered as a subgroup of kind 3 [48, 64]. Some authors have subdivided sort three sufferers into teams, for instance, designating those with progressive myoclonic epilepsy and dementia, and a poorer prognosis, as type 3a [64], but there are clearly unidentified genetic modifiers which decide prognosis [66]. When untreated, death in type 3 sufferers characteristically occurs in childhood or adolescence from pulmonary or hepatic disease and in those with progressive myoclonic epilepsy, neurologic deterioration may trigger demise in maturity despite treatment. It appears that neuropathic Gaucher disease is a continuum [67] from severe neonatal phenotypes to adults with oculomotor issues. Type 2 Infants with acute neuronopathic Gaucher disease appear normal at birth, although splenomegaly may be discovered in the first three months, they usually normally develop some early milestones. Early evidence of neurologic illness could additionally be unusual irritability, a lack of alertness, apparent weakness in holding up the top, oculomotor apraxia, or a exhausting and fast strabismus [24, 50]. Neurodegenerative disease seems in six months and proceeds rapidly to a traditional picture of spasticity and opisthotonus, with trismus, strabismus, and hyperextension of the neck [50]. Death normally outcomes from apnea, aspiration pneumonia, or respiratory failure at a median age of 18 months. The perinatal lethal disease has often been thought of a subtype of infants with kind 2 Gaucher illness in infants with a fast fulminant neonatal onset course [50]. This disorder is reminiscent of the disease in mice homozygous for a null glucocerebrosidase allele who die inside 24 hours [51]. Infants with Gaucher disease might current with lamellar ichthyosis, which can take the form of the collodion baby [52, 53].
100 mg topamax cheap otc
Hyperornithinemia hyperammonemia and homocitrullinuria associated with decreased carbamyl phosphate synthetase I exercise symptoms week by week effective topamax 200 mg. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome: low creatine excretion and effect of citrulline medicine 0829085 purchase topamax 100 mg online, arginine, or ornithine supplement. The mechanism of hyperammonemia and hyperornithinaemia within the syndrome of hyperornithinaemia hyperammonemia and homocitrullinuria. Identification and purification of the ornithine/citrulline service from rat liver mitochondria. A new household affected by the syndrome of hyperornithinaemia hyperammonaemia and homocitrullinuria. Defective ornithine metabolism in cultured skin fibroblasts from patients with the syndrome of hyperornithinemia hyperammonemia and homocitrullinuria. Clinical, biochemical and ultrastructrural examine on the pathogenesis of hyperornithinemia-hyperammonemia-homocitrullinuria syndrome. Hyperornithinemia-hyperammonemia-homocitrullinuria syndrome with stroke-like imaging presentation: clinical, biochemical and molecular evaluation. Gyrate atrophy of the retina: inborn error of l-ornithine: 2-oxoacid aminotransferase. Ornithine-ketoacid aminotransferase in human liver as regards to patients with hyperornithinaemia and familial protein intolerance. The illness is prevalent in Finland, where it has been estimated to happen in a single in 60,000 [2], and Finns or Finnish Lapps have comprised almost half of the sufferers reported [1�5]. The abnormality is in the efflux of those amino acids and could be demonstrated in cultured fibroblasts [10, 11]. Mutations in other populations are various, and more than 50 mutations have been found worldwide [14, 15]. A dry scaly rash is typically seen, in addition to sores on the perimeters of the mouth [16]. A five-year-old boy with continual diarrhea and pitting edema of the decrease extremities was thought to have celiac illness because villous atrophy was found on intestinal biopsy [16]. Treatment of the failure to thrive with parenteral alimentation led to hyperammonemic coma. Increased plasma concentrations of ldl cholesterol and triglycerides are regularly increased [17]. Two-thirds have had fractures, usually of long bones or vertebral compression, and often after minimal trauma [19, 20]. Metabolism of calcium and phosphate is normal, but hydroxyproline excretion within the urine is elevated. These might start in neonatal infancy or be delayed even till adult life in sufferers with welldeveloped aversion to protein [5, 21]. The avoidance of protein-containing meals is an early attribute, which can start as early as 12 months of age. Nonprogressive, asymptomatic opacities have been observed within the lens of the attention [25]. A group of sufferers, from southern Italy, in which consanguinity was excessive, has been described [26] with Genetics and pathogenesis 255 uncommon complications. Manifestations in the patients included abnormalities of the bone marrow, in five of the six examined, by which massive cells resembling seablue histiocytes advised a analysis of Niemann-Pick illness, however sphingomyelinase was unfavorable, and there was additionally prominent erythrophagocytosis. Erythrophagocytosis and immunologic abnormalities have been additionally described in another patient with this disease [27]. As there was additionally high increases of serum ranges of ferritin, lactate dehydrogenase, cytokines as properly as soluble interleukin-2 receptor, Duval et al. This hematologic picture has been seen along with the pancytopenia of propionic acidemia [29]. It has also been observed in carnitine palmitoyl transferase I deficiency [30] and in hemochromatosis [31]. Two sufferers had scientific pancreatitis [26]; in one who required surgical procedure, pancreatic fibrosis was discovered, indicating chronic pancreatitis, as nicely as acute liponecrosis. Some of these patients have presented first in grownup life with interstitial disease of the lung or with renal disease. Heterogeneity of pulmonary displays has been the rule even within a single household. Imaging of the lungs revealed thickened intrabobular septa and tissues, in addition to cysts and ground glass opacity. High-resolution computed tomography has been beneficial for the detection of these abnormalities [33]. Cough, dyspnea, and hemoptysis might occur, and there could additionally be intermittent fever or pulmonary infection. Biopsy of the lung might show cholesterol crystals or granulomas [33] or alveolar proteinosis. Elevated concentrations of cationic amino acids in bronchoalveolar lavage fluid suggest that transport may be irregular in pulmonary epithelium [34]. In numerous sufferers with no pulmonary signs, there was roentgenographic evidence of pulmonary fibrosis [32]. In a examine on 39 Finnish sufferers, proteinuria and hematuria had been noticed in 74 per cent and 38 p.c, respectively. Elevated blood stress, gentle to reasonable renal insufficiency and, in some circumstances, end-stage renal illness and persistent renal failure have been reported [35], with proteinuria and progressive glomerular and tubular insufficiency [26, 35]. Disease in preserving with systemic lupus erythematosus has been reported [36], and the pulmonary disease may be a manifestation of immune advanced problems. Calculation of the renal clearances of cationic amino acids (lysine, arginine, and ornithine) helps to clarify the urinary loss of these amino acids. Oral loading with lysine or arginine to check intestinal absorption could additionally be required for prognosis in such a affected person. Pathology was that of intensive fatty degeneration and micronodular cirrhosis [38, 39]. In different patients, biopsy of the liver has been normal [3, 11] or there have been fat droplets in hepatocyte cytoplasm. At post-mortem within the adult, changes were noted in the glomerular basement membrane, and immunofluorescence positive for IgA indicated an energetic glomerular lesion [39]. Terminal micronodular cirrhosis and pulmonary alveolar proteinosis had been additionally found at post-mortem. Many examples have been discovered of a quantity of affected people in a household [21, 22, 24, 40]. The defect in amino acid transport is usually first evident in the evaluation of amino acids within the blood. The imply focus of lysine in 20 sufferers [37] was 70 mmol/L; those of ornithine and arginine were 21 and 27 mmol/L, respectively. The prognosis may not be clear from these levels, as a result of the conventional ranges may be that low. The mean was 232 mmol/L [37], which is about 4 times the upper limit of regular.
HEP-30 (Chitosan). Topamax.
- Treating periodontitis, a dental condition.
- What other names is Chitosan known by?
- Helping to remake tissue after plastic surgery.
- What is Chitosan?
- Patients with kidney failure who are on chronic hemodialysis. When ingested by these patients, chitosan may reduce high cholesterol; improve anemia; and improve physical strength, appetite, and sleep.
- How does Chitosan work?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96617
100 mg topamax cheap visa
However symptoms before period topamax 100 mg sale, it typically is inconceivable to distinguish carriers from sufferers on the idea of activity of alkaline phosphatase symptoms stiff neck buy generic topamax 200 mg. The traditional measures are employed for the management of fractures and skeletal deformities. Nonsteroidal anti-inflammatory agents may be helpful for bone pain and stress fractures. Bone marrow transplantation has been without effect in severe perinatal hypophosphatasia, however transplantation of allogeneic mesenchymal stem cells cultured underneath osteogenic circumstances and osteogenic constructs made by rising cells on porous hydroxyapatite ceramic yielded proof of improved mineral density of bone and de novo bone formation around the constructs [58]. Enzyme substitute therapy has been under clinical investigation in infants and children. The human gene was bioengineered by extending the C terminus and addition of a deca-aspartate sequence to target mineralizing tissue. Clinical trials reported to date have proven improvement in roentgenographic look, motor operate, power, and agility [60]. Different missense mutations at the tissue-nonspecific alkaline phosphatase gene locus in autosomal recessively inherited types of delicate and severe hypophosphatasia. Identification of novel missense mutations Phe310Leu and Gly439Arg) in a neonatal case of hypophosphatasia. Analysis of localization of mutated tissue-nonspecific alkaline phosphatase proteins related to neonatal hypophosphatasia utilizing green fluorescent protein chimeras. Glu274Lys/Gly309Arg mutation of the tissue-nonspecific alkaline phosphatase gene in neonatal hypophosphatasia associated with convulsions. Rickets, deficiency of "alkaline" phosphatase exercise and premature loss of teeth in childhood. Childhood hypophosphatasia and the untimely loss of tooth: A clinical and laboratory research of seven circumstances. Hypophosphatasia: Screening and household investigations in an endogamous Hungarian village. Hypophosphatasia related to calcium pyrophosphate dihydrate deposits in cartilage. Adult hypophosphatasia with chondrocalcinosis and arthropathy: Variable penetrance of hypophosphatasemia in a big Oklahoma kindred. Recurrent calcific periarthritis, erosive osteoarthritis and hypophosphatasia: A family study. The urinary excretion of phosphoethanolamine in illnesses apart from hypophosphatasia. Inorganic pyrophosphate in plasma in normal persons and in patients with hypophosphatasia, osteogenesis imperfect, and other disorders of bone. Prenatal analysis of hypophosphatasia: genetics, biochemical, and clinical research. Molecular examine of three instances of odontohypophosphatasia ensuing from heterozygosity for mutations in the tissue non-specific alkaline phosphatase gene. Hypophosphatasia: the mutations in the tissuenonspecific alkaline phosphatase gene. New bone formation of allogeneic mesenchymal stem cell transplantation in a affected person with perinatal hypophosphatasia. Hypophosphatasia in kids: Enzyme replacement remedy using bone-targeted, tissue-nonspecific alkaline phosphatase. Mild hyperammonemia and hepatic encephalopathy have been additionally transiently noticed in some people. Hypoketotic hypoglycemia may be present at the side of dicarboxylic aciduria, the latter with no disease locus particular pattern ([8] and private remark G. Detailed investigations of fatty acid oxidation in vivo were all the time normal [2, 3, 6]. In the first years of life, youngsters are very susceptible to infections and crises can occur with each sickness and even vaccinations. Some mother and father, subsequently, kept their youngsters from going to kindergarten faculty within the first years. Prompt intervention by early antipyretic remedy, glucose infusion, and parenteral lipid can ameliorate and shorten the crises. Some individuals had episodes of hypoglycemia, principally related to liver crises but also in a interval following an acute liver crisis. It has remained unresolved, whether or not this can be explained by unspecifically hindered liver operate or whether other mechanisms are responsible. The symptoms are listed based on the frequency of their report in published sufferers, taking collectively 19 patients from 16 households [3, 7, 8]. All these 19 patients have childish hepatopathy, and in all however one, onset was earlier than two years of age. Short stature and dysmorphic features are probably the most frequent extrahepatic findings. Apart from extreme development retardation, she has bilateral optic atrophy with a visual acuity of 0. Two sufferers have been discovered to have small cervical vertebrae (C1, C2) inflicting cervical instability [8]. Large fontanels with delayed closure, quick neck, and abnormal thoracic configuration have also been described. There may be frequent or spontaneous fractures, even from the neonatal age on [5]. Reduced periorbital fats results in the aspect of proptosis in some people and some have a distinguished brow. However, in lots of patients, the physical look is unsuspicious and even utterly normal [3]. Most patients reported have regular intelligence [1, 3, 8], nevertheless, some people had developmental delay and muscular hypotonia [5], and two subjects had epilepsy [3]. She has a outstanding forehead and lowered periorbital fats gives the impression of proptosis. Pelger-Hu�t anomaly, which is characterised by a hypolobulation of granulocytes, was present in all affected Yakut people, but solely in a few of the other sufferers [1, three, 8]. There may be hypogammaglobulinemia and lowered natural killer cells, which could be associated with frequent infections [3, 8]. All of the affected Yakuts had optic nerve atrophy with a imply visible acuity of 0. All people carry at least one missense mutation or a deletion of a single amino acid on one allele. Leu1055Pro), have every been present in three unrelated individuals, while three mutations have every been described in two unrelated sufferers: c. Fixation surgery may turn out to be necessary in case of cervical instability due to hypoplastic cervical vertebrae [8]. In sufferers with hypogammaglobulinemia, immunoglobulin substitute ought to be thought-about. This can cut back frequency of infections, hereby avoiding triggers of liver crises [8]. Neuroblastoma amplified sequence gene is related to a novel quick stature syndrome characterised by optic nerve atrophy and Pelger-Huet anomaly.
Order 200 mg topamax free shipping
The size of the xanthine pool in such a patient is much like symptoms ruptured spleen 200 mg topamax mastercard that of hypoxanthine medicine 802 100 mg topamax discount overnight delivery, and its turnover is about one-third that of hypoxanthine, however roughly eighty percent of the xanthine turned over every day seems within the urine, whereas only 5 p.c of that of hypoxanthine seems in the urine. These metabolic features are important for the affected person due to the insolubility of xanthine (Table seventy two. By contrast with xanthine, uric acid is relatively soluble in human urine at pH values which are achievable. For these causes within the presence of xanthinuria the formation of calculi is to be expected. Allopurinol is effective within the treatment of gout and myeloproliferative disease by virtue of its effectiveness as an inhibitor of xanthine oxidase [15]. Reported exercise has ranged from undetectable to lower than 10 % of control [3�5, 16�18]. In another Japanese man discovered to have hypouricemia in a health check-up, a 445C > T transition was present in exon 6 [20]. Xanthinuric sufferers unable to convert allopurinol to oxypurinol also had poor activity of aldehyde oxidase. A xanthinuric patient with normal formations of oxypurinol had normal aldehyde oxidase activity [24]. Urinary excretion of inosine has been reported to be elevated in sufferers with xanthinuria [25]. The formation of xanthine is derived largely from guanine nucleotide degeneration under basal conditions and after intravenous fructose [25]. This would explain the prominence of xanthine of the oxypurines in hereditary xanthinuria. The relative insolubility of xanthine might make fluid consumption irrelevant; it might nonetheless appear prudent to eat loads of fluids. In a patient with residual xanthine oxidase exercise, allopurinol could probably be efficient in changing a variety of the oxypurine excreted to the soluble hypoxanthine. Allopurinol is efficient in the remedy of gout and myeloproliferative illnesses, by virtue of its effectiveness as an inhibitor of xanthine oxidase [25]. Identification of two mutations in human xanthine dehydrogenase gene responsible for classical sort I xanthinuria. Hereditary xanthinuria with extreme urolithiasis occurring in infancy as renal tubular acidosis and hypercalciuria. Clinical, physiological and biochemical research of a affected person with xanthinuria and pheochromocytoma. The mass-spectrometric identification of hypoxanthine and xanthine ("oxypurines") in skeletal muscle from two sufferers with congenital xanthine oxidase deficiency (xanthinuria). Quantitation of oxypurines and allopurinol metabolites in biological fluids by cation-exchange chromatography. The effectiveness of a xanthine oxidase inhibitor allopurinol in the remedy of gout. Identification of a new level mutation in the human xanthine dehydrogenase gene liable for a case of classical kind I xanthinuria. Xanthine dehydrogenase deficiency with novel sequence variations presenting as rheumatoid arthritis in a 78-year-old affected person. Metabolism of pyrazinamide and allopurinol in hereditary xanthine oxidase deficiency. Demonstration of a mixed deficiency of xanthine oxidase and aldehyde oxidase in xanthinuric patients not forming oxypurinol. The rarity of the disease is indicated by the truth that it was 1965 earlier than a second affected person was described [2]. The importance of those two papers is reflected not solely in the thoroughness of the medical and metabolic documentation, but by the fact that they set out the definitive remedy with uridine [2, 3] which has been terribly efficient and serves as a model for the reality that the thorough understanding of the nature of a elementary defect can result in rational and efficient remedy. The gene on chromosome 3 has been sequenced, and a small number of mutations has been defined [6]. Patients have just lately been reported by which orotic aciduria was not associated with megaloblastic anemia [7, 8]. They were thought-about to have a particular kind of orotic aciduria in which the ratio of orotic acid to orotidine approximated 1(range 0. A few younger infants offered with anemia in which growth had to date (two months) been normal [12]. A seven-year-old who developed symptoms of anemia at six years of age, but who had apparently been pale previously, had normal development and intellectual growth [13]. One affected person treated at 19 months with uridine had not had his nails trimmed for the earlier six months [2]. Hemoglobin levels have usually been 7�8 g/dL, hematocrit approximating 25 p.c, but some have had extra severe anemia [14]. Red cell morphology has been unusual, with a marked diploma of anisocytosis and poikilocytosis [2], macrocytosis and a lot of strikingly massive and oval shapes with long diameters. Many macrocytes had been hypochromic, while ranges of iron are normal, or elevated [5]. Occasional polychromatic cells have been seen, in addition to strippled cells, Howell�Jolly our bodies, Cabot rings, and nucleated erythrocytes. Bone marrow aspirates reveal megaloblastic changes in a majority of the nucleated pink cells. Concentrations of B12 and folic acid are regular, and early patients had been treated with B12 and folate without effect [1, 2]. Gastric aspirates did present achlorhydria, a few of them responsive to histamine [2, 15] and some not [2]. The urine seems normal as handed, however on standing, particularly within the chilly, a large, white precipitate varieties [1, 2, 12, 15]. The disease was identified as an inborn error of metabolism by the recruitment by Huguley et al. One patient [13] got here to consideration because of hematuria and was discovered to be anemic. Oliguria might accompany infection, or decreased fluid consumption or dehydration from some other supply such as diarrhea [1]. Under such circumstances, there may be urinary tract obstruction due to orotic acid sludging within the ureters or urethra [1]. An intravenous pyelogram revealed a nephrogram effect with radiopaque materials remaining in the kidney 1. Urea nitrogen and creatinine in blood are often normal however will rise in the presence of obstruction. An unusual function was deficiency of specific immunoglobulins in a single affected person [12].
Buy 100 mg topamax overnight delivery
Diagnosis has generally required the assay of the enzyme in biopsied liver medicine 5000 increase topamax 200 mg with mastercard, but mutational analysis might allow its avoidance medicine 60 generic topamax 200 mg amex. Culture of monocytes in media wealthy in calcitriol has been reported [32] to permit the analysis by enzyme assay. A radiochemical methodology has developed that detects faulty (absent) enzyme exercise in isolated monocytes earlier than and after incubation with calcitriol in tissue tradition media [33]. The insG960-961 accounted for forty six percent of twenty-two mutant alleles in Japanese and 14 of 28 mutant alleles in non-Japanese [7]. The fasting that ordinary infants often bear within the first days of life makes for a neonatal presentation in sufferers with little residual enzyme exercise [35]. If examined with a provocative quick, patients turn into hypoglycemic when saved glycogen is exhausted. This might occur in a quantity of hours in an toddler [15] or after 14�20 hours in an older affected person [1, 2, 15, 34]. Depletion of glycogen previously synthesized from glucose by injection of glucagon early within the quick ensures that gluconeogenesis is being tested adequately (Chapter 49) [10]. Administration of glucagon after the event of hypoglycemia leads to no glycemic response. Hypoglycemia can also be generated by a food plan low in carbohydrate and excessive in protein and fats [10]. This check has been employed in sufferers judged on the premise of the response to fasting to have lactic acidemia, due to a defect in gluconeogenesis (Chapter 49), to have the ability to suggest that the enzyme be assayed on biopsied liver. Following fructose, the blood sugar drops to hypoglycemic ranges inside 15 minutes; lactic acidemia and increased levels of alanine and uric acid accompany systemic acidosis. Glycerol loading results in a response just like that with fructose [1, 7, 30, 38]. Rather, the individual tolerance of the patient can be explored cautiously [14], while delicate drinks that provide a sucrose or fructose load must be prevented. Lists can be found [37] that provide the sugar content material and its nature of medicinal liquids. Patients, families, and physicians should significantly be warned about antibiotic elixirs or tylenol syrup which tend to be prescribed when the affected person is already metabolically compromised with infection and vomiting-related fasting. A snack at bedtime is usually helpful, as is uncooked cornstarch (Chapters 39 and 59). One affected person developed gout at 32 years of age and was treated with allopurinol [33]. Fasting hypoglycemia and metabolic acidosis related to deficiency of hepatic fructose-1,6diphosphatase exercise. A baby with lactic acidemia and fructose-1,6-diphosphatase deficiency within the liver. Identification of genetic mutations in Japanese sufferers with fructose-1,6bisphosphatase deficiency. Biochemical and clinical observations in 4 sufferers with fructose-1,6-diphosphatase deficiency. Acidose lactique hypoglyc�mie et h�patom�galie par deficit h�r�ditaire en fructose-1,6-diphosphatase h�patique. Recurrent attacks of ketotic acidosis related to fructo-1,6-diphosphatase deficiency. Deficiency of glucose6-phosphate dehydrogenase present in a case of hepatic fructose-1,6-diphosphatase deficiency. Detection of heterozygotes for fructose-1,6-diphosphatase deficiency by measuring fructose-1,6-diphosphatase exercise of their cultured peripheral lymphocytes. Urinary sugar phosphates and associated organic acids in fructose-1,6-diphosphatase deficiency. Fructose and glucagon loading in siblings with fructose-1,6-diphosphatase deficiency in fed state. Fructose-1,6diphosphatase deficiency: analysis utilizing leukocytes and detection of heterozygotes with radiochemical and spectrophotometric technique. Experience of King Faisal Specialist Hospital and Research Center with Saudi natural acid problems. Diagnosis of fructose1,6-bisphosphatase deficiency utilizing cultured lymphocyte fraction: a secure and noninvasive alternative to liver biopsy. Fructose 1,6-bisphosphatase deficiency: enzyme and mutation evaluation carried out on calcitriol-stimulated monocytes with a observe on long-term prognosis. Hypoglycemia and lactic acidosis related to fructose-1,6-diphosphatase deficiency. There are eight different protein parts, in seven of which human deficiency illness has been documented. Insulin also activates the enzyme, by fostering the prevalence of the dephosphorylated form. Because of gluconeogenesis, the amount of carbohydrate supplied to peripheral tissues from the liver exceeds the quantity ingested. The mind is a major site of its oxidation, and this proportion is larger in infancy and childhood due to the higher proportion of the brain measurement to body measurement. The dependency of the mind on oxidative metabolism makes it significantly prone to harm in ailments of oxidation that result in lactic acidosis. Most of those in males have been missense mutations, whereas in females there have been major disruptions in the single affected X chromosome [21, 22]. In a collection of fifty four patients with deficiency of E1, the spectrum of medical presentation was as broad as those of the lactic acidemias in general (Chapter 47). The majority of these infants die before six months of age, lots of them within the neonatal interval [25�28]. There is a somewhat extra indolent presentation in patients with continual, more modest lactic acidemia who first come to consideration due to delayed psychomotor growth [29�36]. Many of these sufferers have the clinical and neuropathologic options of Leigh syndrome [37]. They could expertise speedy deterioration or episodic deterioration following infections. A number of seizures includes grand mal, myoclonic, absence, or akinetic convulsions. Neuroimaging could reveal attenuated signal in the basal ganglia, notably in putamen and globus pallidus [38, 39], and in the end generalized cerebral atrophy. Histopathologic examination reveals spongiform degeneration and gliosis especially within the basal ganglia. Among the neonatal acidosis group, some have had cortical cysts at autopsy [24, 39].
Generic topamax 200 mg on-line
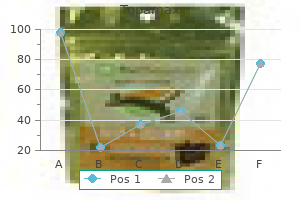
Prenatal prognosis has been completed by the ultrasonographic appearance of the fetal head at sixteen weeks [47] symptoms 6 days post embryo transfer 100 mg topamax buy free shipping. Prenatal analysis can additionally be accomplished by the measurement of alkaline phosphatase exercise in cultured amniocytes [48] medicine remix order 200 mg topamax visa. Family research indicate clearly that the different clinical phenotypes symbolize different disease entities. Certainly, the severe neonatal illness and the mild juvenile illness by no means occur collectively in a kindred [27]. A type of hypophosphatasia has been reported [49] during which transmission is autosomal dominant. Clinical features were untimely loss of enamel, bowed legs, and a beaten silver look to the roentgenogram of the skull. It is evident that the classic delicate hypophosphatasia, including asymptomatic individuals who have phosphoethanolaminuria and a few with odontohypophosphatasia have dominant expression of mutation on a single allele [7, 50�52]. E174K mutation is comparatively common in Caucasians, occurring in 31 p.c of individuals with delicate hypophosphatasia [56]. The heterozygous state can normally be demonstrated by measuring the serum activity of alkaline phosphatase [3]. Developmental delay, dysmorphic options, neonatal spontaneous fractures, wrinkled skin, and hepatic failure: a brand new metabolic syndrome Identification of the neuroblastoma-amplified gene product as a component of the syntaxin 18 advanced implicated in Golgi-to-endoplasmic reticulum retrograde transport. Patients should be equipped with an emergency card, letter, or bracelet containing instructions for emergency measures and telephone numbers. The protein itself had been isolated in 1955 by Shultze and colleagues [3], who recognized its function as the major trypsin inhibitor of serum and the most important part (90 percent) of the 1-globulin fraction [4]. It was evident early that partial deficiency was transmitted in an autosomal dominant manner, and that these with extreme deficiency had been homozygotes [5]. The present method of alternative is isoelectric focusing on polyacrylamide gel [7], and seventy five variants have been acknowledged. The disease offers a model for the understanding of the processing of accurately and incorrectly folded glycoproteins within the endoplasmic reticulum [11]. The relationship of protease and elastin has led to understanding of the pathogenesis of emphysema not solely on this widespread illness, but in different nongenetic forms of emphysema. These 808 1-Antitrypsin deficiency patients developed particular manifestations of liver disease within the first year of life with early cholestasis that resolved by 6 months of life, however elevated serum ranges of hepatocellular enzymes and hepatomegaly continued. The most frequent hepatic presentation is with a neonatal hepatitis syndrome [22]. Bilirubinemia is a uniform indicator of cholestatic disease as a result of indirect bilirubin is sure to albumin and not present in urine. Occasionally in such an toddler, there are acholic stools and the picture could simulate extrahepatic biliary atresia [25]. Another presentation is with transient signs of liver disease occurring with intermittent infection or appendicitis at 2 years or later in a beforehand asymptomatic child [26]. In one other group of sufferers, hepatomegaly has been found in childhood and not utilizing a historical past of neonatal jaundice. A quantity have died of this or other complications of cirrhosis during childhood or adolescence, even in infancy, and certainly after the development of cirrhosis. This is the so-called pseudochylous ascites by which some sufferers with chronic liver illness have lactescent fluid. She had been found to have hepatomegaly at 2 years-of-age and biopsy revealed cirrhosis. She had two spider telangiectases on the arms and a large healed incision over the liver, which was palpable three cm beneath the costal margin. Protein content material has been used to distinguish exudative from transudative processes, but the range of protein content in liver disease is simply too nice to permit this distinction. Coagulation elements could additionally be reduced, and there may be clinical bleeding, particularly gastrointestinal. Cirrhosis has additionally been observed in a selection of adults who had a historical past of neonatal hepatitis or liver illness in childhood [28]. It is classically early in onset, occurring at 20�40 years of age in smokers and fifty five in nonsmokers [5, 30, 31]. Cough develops in about half of the sufferers, and recurrent pulmonary infections are widespread. On examination, the patient could also be thin, but the diameter of the chest is increased. Breath sounds are diminished, and the chest film reveals hyperinflation, especially in the bases. Pulmonary perform tests are typical of severe emphysema consistent with a lack of pulmonary elastic recoil. Air move is proscribed, and diffusion capability and most transpulmonary pressure are lowered. Hypocarbia and respiratory alkalosis could additionally be related to delicate pulmonary hypertension. Electrocardiograms might show chronic pressure on the right coronary heart with proper axis deviation and proper atrial hypertrophy. There have been no pulmonary symptoms, but bronchial markings have been increased as linear densities, there was hilar prominence, and there was proof of hyperinflation. Smoking influences not solely the age of onset of emphysema however the rate of its development [35�38]. The pathology of the lung has indicated that emphysema outcomes from a damaging process involving the alveoli. Electron microscopy reveals intensive destruction of alveolar septal walls with loss of alveolar construction and huge air-filled spaces. The pathology of the liver offered early insights into the molecular pathogenesis of the disease. In addition, the adjustments of chronic hepatic disease are nonspecific, but progressive. In sufferers with cystic fibrosis, the Z phenotype is a risk factor for the event of extreme liver illness with cirrhosis and portal hypertension [41]. In addition to illness of the lungs, membranous glomerulonephritis has been noticed histopathologically, and some of these patients have had signs of renal dysfunction [42]. Some proof of glomerular disease is common at post-mortem examination in sufferers dying of liver disease. A variety of inflammatory disorders have been associated with heterozygosity for the Z allele, together with rheumatoid arthritis [43] and uveitis [44]. Its molecular weight is fifty two kDa, and its carbohydrate content material of 12 percent contains a variety of sialic acid residues. The protein synthesized in the liver is longer, containing a signal peptide and an N-terminal methionine [51].
Discount topamax 200 mg on-line
The glucose-6-phosphatase catalytic protein is regular and may be assayed if membranous elements of the liver cell are disrupted by freezing or remedy with detergents medications during labor discount 100 mg topamax mastercard, but in situ the system is nonfunctional [3 medicine list buy topamax 100 mg on-line, 78, 79]. The defect can be demonstrable in leukocytes, which have impaired uptake of glucose [80] in type Ib, and this will present a method to check for the dysfunction. Among patients with partial deficiencies, some have decreased ranges of immunoprotein and others normal amounts [14, eighty two, 83]. The gene for human hepatic glucose-6-phosophatase on chromosome 17q21 has been cloned, and a selection of mutations have been recognized [84]. Of the 2 Japanese sufferers with kind Ib without neutropenia, one had R415X, which had previously been encountered in sufferers with neutropenia, on one allele and G339D on the other. The different affected person was homozygous for G794A, which led to a splicing error, deleting exon 3. In a study of adenomas in glycogenosis Ia, alterations in chromosome 6, similar to a gain of 6p and a lack of 6q have been outstanding [90]. Infections are notably dangerous, and the patient could require admission to hospital and therapy with parenteral fluids containing glucose and electrolytes. There is a distinct tendency for enchancment with age, even by the age of 4 or 5 years [17, 30]. By the time of puberty, considerable amelioration has often been observed [12]; the enlarged liver takes up significantly less of the stomach and hypoglycemic symptoms are much less outstanding. However, little improvement in long-term prognosis because of therapy occurred until lately. Portacaval shunting has primarily been abandoned within the remedy of this disease, nevertheless it was famous that the parenteral alimentation attendant on the procedure led to discount in hepatic dimension and reversal of metabolic abnormalities, the expansion failure and the bleeding diathesis [91, 92]. These observations targeted consideration on approaches to the extra common provision of glucose to meet tissue wants. The approaches which have been successful are continuous nocturnal nasogastric or gastrostomy feeding [92�95] and oral uncooked cornstarch [95, 96]. With either regimen, frequent high carbohydrate meals by which 65�70 % of the energy are carbohydrate are employed through the day. Dietary consumption of fructose and galactose is restricted in some centers and never in others. The optimum quantity for every affected person is determined individually; passable outcomes have been confirmed with 1 g/kg each six hours. Older patients could not require a feeding in the midst of the night if bigger quantities (2�4 g/kg) may be taken at bedtime. This regimen has been proven to keep euglycemia and to reverse scientific and biochemical disturbances in most sufferers [97, 98]. Some begin at six months and some at 12 months, using maltodextrin previous to that. Glucose, Polycose, and elemental formulations have been employed, each providing 8�10 mg/kg per minute in an infant and 5�7 mg/kg per minute in an older baby. There is a bent towards the development of hypoglycemia in the morning after the nocturnal feeding is stopped; so that the first meal ought to be within 15�30 minutes of discontinuing the nocturnal feeding. Hypoglycemia and dying have been reported following malfunction of the pump or dislodging the tube [101]. Some sufferers have required a combination of cornstarch and nocturnal nasogastric feedings. Patients with glycogenosis kind Iasp and Ib must also be managed with these regimens. In sort Ib, granulocyte and granulocyte�macrophage colonystimulating factors have been employed to combat the neutropenia and deal with the inflammatory disease [102]. Both regimens have been employed long sufficient to have provided [103] encouraging long-term effects on the course of the illness [97, 98, 103, 104]. Surprisingly, therapy has been reported to be related to an absence of development of hepatic adenomas [100] and regression of adenomas has been noticed. Guidelines for the management of glycogenosis sort I are primarily based on consensus of the European research [105]. Overall recommendations are for 60�65 percent of total energy from carbohydrates and 10�15 p.c from fat. In our view, the risk of catastrophic hypoglycemia with malfunction of tube or pump is an argument in favor of cornstarch. Angiotensin changing enzyme inhibitors have been beneficial [105] for hypertension. Transplantation of the liver supplies a definitive treatment of the illness [110, 111]; nonetheless, the magnitude of the process has tended to make its use reserved for a small number of sufferers with refractory illness or, after all, hepatic malignancy. A new variant of glycogen storage illness kind 1 most likely because of a defect in the glucose-6-phosphate transport system. Type 1b glycogen storage disease is attributable to a defect within the glucose-6-phosphate translocase of the microsomal glucose-6-phosphatase system. Localisation of the gene for glycogen storage disease type 1c by homozygosity mapping to 11q. Mutations within the glucose 6-phosphatase gene that cause glycogen storage illness type Ia. Sequence of a putative glucose 6-phosphate translocase mutated in glycogen storage disease type Ib. The Fanconi syndrome associated with hepatic glycogenosis and irregular metabolism of galactose. Markedly elevated serum biotinidase exercise could point out glycogen storage illness kind Ia. Functional differentiation of glycogenoses of the liver with respect to the use of glycerol. Hemorrhage into a hepatic adenoma and type Ia glycogen storage illness: a case report and evaluate of the literature. Nephrolithiasis hypocitraturia and a distal renal tubular acidification defect in sort I glycogen storage disease. Diagnosis and management of glycogen storage disease type 1: a follow guideline of the American College of Medical Genetics and Genomics. A new mutation of the glucose-6-phosphatase gene in a 4-year-old lady with oligosymptomatic glycogen storage illness type Ia. Circulating lipids and lipoproteins in glycogen storage illness sort 1 with nocturnal nasogastric feeding. Urinary excretion of C6-C10 dicarboxylic acids in glycogen storage ailments sorts 1 and three. Neutropenia and impaired neutrophil migration in sort 1B glycogen storage disease. Glycogenosis sort Ib difficult by extreme granulocytopenia resembling inherited neutropenia.