

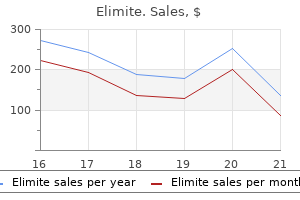
Elimite dosages: 30 gm
Elimite packs: 2 creams, 3 creams, 4 creams, 5 creams, 6 creams, 7 creams, 8 creams, 9 creams, 10 creams

30 gm elimite purchase with visa
The direction of movement of outer root sheath cells is unclear but they might migrate downwards in the path of the hair bulb acne 6 months postpartum generic elimite 30 gm mastercard, the com panion layer forming the plane of slippage between the internal and outer root sheaths skin care 4 less elimite 30 gm order online. The outer root sheath of the suprabulbar region merges imperceptibly with the isthmus the place the innermost cells endure tricholemmal keratinization. The hair cuticle is fashioned initially as a single cell layer, but the cells turn into progressively imbricated (tilelike) as they transfer periph erally. The cells turn out to be flattened, first in a path at proper angles to the aircraft of the follicle, after which becoming progressively angu lated so that the outer edges of the cells level in an upward direc tion. The cuticle has important protective properties: it acts as a barrier to physical and chemical insults, and likewise maintains the integrity of the hair shaft. Eventually this course of might lead to exposure of the cortex and fracture of the hair shaft. Cells destined to form the cortex gradually turn into more fusiform in shape as they migrate upwards from the hair bulb. Keratin filaments are crosslinked to keratinassociated proteins, which kind a matrix between the fil aments. They are classified into three main fami lies: excessive sulphur, ultrahigh sulphur and excessive glycine�tyrosine proteins [33]. The medulla is a variable construction in human hairs, the place it may be steady, discontinuous or absent. A plexus of longitudinally aligned sensory nerve fibres surrounds the isthmus area. Small nerve fibres may be organized in a circular style outside the longitudinal fibres. Several several types of nerve endings are discovered round human hair follicles, together with free nerve endings, piloRuffini nerve finish ings and Merkel nerve endings [35,36]. In other species, lamellated nerve endings are found in richly innervated sinus hair follicles. The comparatively translucent filaments, set in a extra dense sulphurrich matrix, seem as concentric lamellae (macrofibrils), giving a characteristic fingerprint pattern. The interval of energetic hair development is named anagen and the duration of this phase is responsible for figuring out the ultimate size of the hair. In most hair follicles in most animals, anagen is relatively temporary, lasting a few weeks at most. Under normal circumstances, 80�90% of hair follicles on the human scalp are in anagen at anyone time. The entry of a resting hair follicle into anagen is heralded by the onset of mitotic activity in epithelial cells overlying the dermal papilla on the base of the follicle (the secondary epithelial germ). In most follicle sorts (vibrissae follicles are an exception), the decrease a part of the follicle elongates downwards alongside a preformed der mal tract (the stele). The dermal papilla expands from a tightly packed ball of cells into a flask formed structure the place the cells turn into separated by an extra mobile matrix rich in proteoglycans and basement membrane proteins. A network of capillary blood vessels develops around the lengthening follicle, extending into the dermal papilla in bigger follicles. In the absolutely developed anagen follicle, epithelial cells within the hair bulb bear vigorous proliferative exercise. Their prog eny transfer distally and differentiate in an ordered style to form the layers of the inside root sheath and the hair shaft. At the top of anagen, epithelial cell division declines and ceases, and the follicle enters an involutionary section often identified as catagen. The base of the follicle, together with its dermal papilla, strikes upwards, ultimately to lie just under the level of the arrector insertion. The interval between the com pletion of follicular regression and the onset of the subsequent anagen section is termed telogen. In the human scalp, hair follicles may stay in a state of latency, also called kenogen, for a pro longed period after the club hair is shed [37]. Control of the hair cycle Hair cycling is managed primarily within individual hair follicles however this intrinsic behaviour may be modulated by each native and systemic elements. There is a distinguished glassy membrane surrounding the regressing epithelial column. Moult waves are regulated throughout the pores and skin and are accompanied by modifications in different skin constructions, corresponding to epidermal and dermal thickness. In many mammals, living in their natural setting in mood ate and higher latitudes, moult waves continue into grownup life and occur on a seasonal basis. Seasonal moulting is regulated by the endocrine system under the affect of environmental indicators. The pro duction of melatonin by the pineal gland, which transduces visual alerts, and prolactin by the pituitary, play a key position in orches trating endocrine management of seasonal hair progress [40�42]. Vestiges of seasonal variation in hair development are current in humans [43], although the magnitude is seldom adequate to be noticeable. Although local and systemic components modulate the hair cycle in some species, in grownup people and some other mammals hair cycling is asynchronous. In people, for example, the dur ation of anagen on the scalp may last a quantity of years, whereas on the eyebrows anagen is very transient. When scalp hair follicles are transplanted into different regions of the skin they preserve the cyclical behaviour of the donor site, indicating that cycle management is deter mined inside the follicle or its instant tissue environment. Interactions between two key cell populations in the hair follicle � epithelial stem cells in the outer root sheath and mesenchymal cells within the dermal papilla and dermal sheath � are thought to underlie intrinsic control of hair cycling, and a lot of molecules have been implicated in this process [44]. Fluctuations within the inhibitory influence of bone morphogenetic proteins and the stimulatory Wnt/catenin pathway appear to play a key role in regulating stem cell exercise through the hair cycle. First, they par ticipate within the endocrine management of moulting in animals that show seasonal hair development [46]. Second, in some mammals, androgens stimulate the expansion of hair follicles in certain areas of the skin fol lowing sexual maturity. Third, in people and another primates, androgens are essential for the development of balding on the scalp. The progress of apparent facial, trunk and extremity hair within the male, and of pubic and axillary hair in each sexes, depends hair growth 89. The growth of such hair at puberty is, in broad phrases and a minimum of initially, in parallel with the rise in ranges of androgen from testicular, adrenocortical and ovarian sources, which happens in both sexes and is considerably steeper in males. That testosterone from the interstitial cells of the testis is answerable for the expansion of beard and body hair in male adolescence and that testicular exercise is itself initiated by gonadotrophic hor mones of the pituitary is unquestioned. However, the findings that progress hormonedeficient boys and girls are less than normally aware of androgens, and that development hormone is important as a synergistic issue to allow testosterone to be fully efficient with respect to hair development [47], as properly as protein anabolism and growth promotion, counsel that hypophysial hormones also have a extra direct function. Direct proof of the role of testicular andro gen is that castration reduces progress of the human beard [48], whereas testosterone stimulates it in eunuchs and aged men.
Elimite 30 gm generic otc
Amiodarone: generally acne location meaning discount elimite 30 gm amex, the pigmentation is slowly reversible and fades over months to years after discontinuation of the drug skin care uk 30 gm elimite cheap free shipping. Dose discount or withdrawal of amiodarone can result in complete disappearance of the pigmentation. Antimalarial medicine: discoloration slowly fades after discontinuation of remedy, however might take a quantity of months to disappear, and barely resolves completely. With continued therapy, the areas darken, notably oval patches on the shins, which enhance in dimension. Pathology the slatebrown color in mounted drug eruption is due to pigmentary incontinence with melanophages in the upper dermis [4]. Most regularly reported drugs include tetracyclines, nonsteroidal antiinflammatory medicine, sulfonamides and sedatives. More or less symmetrical, discrete patches are usually seen however the melanosis could additionally be diffuse or melasmalike, and the mucous membranes could additionally be concerned [6,7]. The genitalia and perianal space are sometimes affected, although the eruption can seem wherever on the skin floor. The characteristic course is recurrence of lesions on the similar sites with development of latest areas of involvement with repeated publicity to the causative agent. Linear, streaky pigmentation following an acute blistering reaction caused by large hogweed and sunlight. Squeezing limes outside when making ready cold drinks can cause blistering of the arms if carried out on sunny days. Berloque dermatitis: the distribution of the lesions is subsequently variable but their configuration is normally distinctive. Pathophysiology Phytophotodermatitis: all of the crops reliably recorded as inducing this response in people have been proven to include furocoumarins: they embody cow parsley (Anthriscus sylvestris) and big hogweed (Heracleum sphondylium) [1,2]. Disease course and prognosis Favourable prognosis if the inflicting agent is averted. Phytophotodermatitis [1�5] and Berloque dermatitis [6�9] are two distinctive clinical syndromes. Phytophotodermatitis: initially, intensely pruritic papulovesicular lesions with irregular shapes and crisscrossing linear streaks could also be present. Pathophysiology Predisposing elements the intensity and persistence of the hypermelanosis are higher in darkskinned subjects. Hypermelanosis commonly follows acute or continual inflammatory processes within the pores and skin. Similarly, in mounted drug eruptions, hyperpigmentation occurs as a end result of injury of cells within the basal layer. This could be defined by an increased mitotic rate of keratinocytes, diminished switch of melanosomes from the melanocyte to keratinocytes and a decreased transit time of the latter from the basal layer to the skin surface. Very frequently in inflammatory pores and skin disease, hypermelanosis and hypomelanosis occur collectively, usually with a slatyblue color due to the presence of melanophages within the upper dermis. The explanation for the pigmentation is normally obvious, though the preceding lesions have generally not been noticed by the affected person or have been transitory or clinically imperceptible. Reticulate pigmentation comparable to the underlying vascular community is seen in erythema ab igne (see Chapter 126), a more lately described reason for which is warmth from laptop computers quite than open fires or sizzling water bottles [6]. Infective causes embrace late secondary syphilis, in which diffuse hypermelanosis of the edges and back of the neck and the shoulders could develop (leukoderma colli syphiliticum) (see Chapter 29) [7] and late pinta during which slatyblue dyspigmentation could 88. Postinflammatory hyperpigmentation may also occur following trauma to the skin, including procedures corresponding to dermabrasion, particularly in darker pores and skin types. Classification of severity the degree of inflammation seems to be of much less significance in determining the pigmentary response than the character of the dermatosis, for it could be frequent and severe after some situations and slight after others. Disease course and prognosis the skin lightens slowly over time spontaneously or with remedy. Pathophysiology the underlying reason for ashy dermatosis is unknown and is likely to be heterogeneous. Those cases by which erythema is current share many features with lichen planus together with lichenoid inflammation histopathologically with basement membrane zone harm and infiltration of T lymphocytes [6,7]. Ultrastructural research present vacuoles inside the cytoplasm of basal and suprabasal keratinocytes that contain many melanosome complexes. It has been proposed that ashy dermatosis be used for all such circumstances however that erythema chronicum perstans be restricted to those instances during which an inflammatory section with erythema has been noticed [1]. In a latest evaluate of 68 patients from Korea, lower than a fifth had been noticed to have peripheral erythematous borders to their lesions [5]. Disease course and prognosis the preliminary erythematous part tends to settle after several months [1]. The macules differ in size and have a tendency to coalesce over in depth areas of the trunk, limbs and face. Against the final greyish background are macules of hypomelanosis or hypermelanosis. The lesions are mostly asymptomatic, although some sufferers could expertise gentle pruritus. Treatment depends primarily on establishing the trigger and if attainable reversing the circumstances that have given rise to the hypermelanosis. The most wellstudied hypopigmenting agent used to treat melasma and other hypermelanotic circumstances is hydroquinone. Topical hydroquinone is mostly used in a focus of 4%, included in chilly cream or a hydroalcoholic base. Acute side effects related to hydroquinone embody rare allergic reactions, postinflammatory hyperpigmentation and transient hypopigmentation [1]. It may be utilized alone or within the broadly used triple mixture cream consisting of hydroquinone 4%, a retinoid and a corticosteroid. Topical hydroquinone or this triple combination cream are highly effective and protected and can be thought-about as first line agents within the therapy of melasma. Because of concern about steroidinduced facial atrophy, telangiectasia and rosacealike acneform eruptions, using triple combination cream has been limited to no more than twice every day for six months, disqualifying it as a upkeep remedy for melasma [1,2]. Topical hydroquinone and tretinoin are also effective within the treatment of postinflammatory hyperpigmentation but require extended remedy [3]. The monobenzylether of hydroquinone has been used only to bleach away the remaining pigmented areas in patients with extensive vitiligo [4]. In instances of pigmentary problems corresponding to melasma which would possibly be refractory to topical treatment, mixture with procedures similar to peels or laser could be thought-about. Chemical peels with glycolic acid or salicylic acid can be a helpful adjunct to those topical remedies, although the therapeutic response is usually unsatisfactory and a universally efficient chemical peeling has not yet been discovered [1]. Laser or light therapy for the therapy of melasma has turn out to be more and more popular. As with chemical peels, they carry an increased danger of side effects due to direct harm to the skin. Despite the risks, some promising outcomes are seen in randomized trials utilizing laser or mild therapy [3,7].
Diseases
- Erythema nodosum
- Oculoauriculofrontonasal syndrome
- Hyperprolinemia
- Mental retardation short stature absent phalanges
- Contractural arachnodactyly
- Enolase deficiency type 3
- Trevor disease
30 gm elimite buy overnight delivery
Avulsion of the limbs could occur during supply due to skin care qualifications 30 gm elimite purchase visa a generalized connective tissue fragility acne zoomed in buy cheap elimite 30 gm online. This form has been subdivided on the idea of rib and limb bone abnormalities [13,16]. Multiple recurrence because of gonadal mosaicism can mimic autosomal recessive inheritance [17]. As the kid grows older, progressive scoliosis and bowing of long bones cause crippling deformities. It is inherited as an autosomal dominant trait, although sporadic instances also happen [9]. The sclerae are blue or gray, and straightforward bruising and earlyonset deafness are frequent, but skeletal deformity is absent or delicate. The incidence of mitral valve prolapse is elevated, and the aortic valves are skinny and occasionally incompetent [10�12]. Disease course and prognosis the prognosis varies relying on the severity of symptoms. Respiratory failure adopted by unintentional trauma are the most typical causes of premature demise. Investigations Prenatal diagnosis of the extra severe types is possible, using ultrasonography from week 16 [19]. A multidisciplinary method, including medical, orthopaedic, physiotherapy and rehabilitation enter, is required with the purpose of Inherited generalized cutis laxa 72. No dependable information has been printed but the prevalence at delivery is estimated at around 1/1000 000. Age Presentation is at birth or early infancy though the autosomal dominant kind has a later and milder onset. Second line Somatic cell therapy, using allogeneic bone marrow and mesenchymal stromal cell transplantation have been used [23,24]. Associated diseases Pathophysiology Cutis laxa is attributable to disruption to regular elastic tissue function. Three major teams of proteins have thus far been identified as answerable for the abnormal elastic fibre morphology. The inherited types embrace autosomal dominant, a number of autosomal recessive sorts and an Xlinked type. Synonyms and inclusions � Generalized elastolysis � Generalized elastorrhexis � Generalized dermatochalasis Pathology the skin is of normal thickness, but the elastic fibres are sparse, brief, fragmented and clumped, significantly in the higher dermis, and they show granular degeneration [5]. Approximately 30% of patients with autosomal dominant cutis laxa have de novo mutations with no household history [8]. Clinical options Introduction and general description Generalized cutis laxa is rare. The medical presentation and the mode of inheritance show considerable heterogeneity. Cutis laxa variably affects connective tissue within the skin as properly as other components of the body, including the guts, blood vessels, joints, intestines and lungs. Abnormal glycosylation of proteins Cutis laxa, progress and developmental delay, joint laxity, microcephaly, triangular face, massive ears, premature aged look, osteoporosis. Lungs not affected Phenotypic overlap with cardiofaciocutaneous syndrome Characteristic coarse facies, quick stature, cutis laxa notably palms and ft, severe feeding problem and failure to thrive, cardiac anomalies, facial warts Often presents in childhood Cutis laxa, development retardation, dysmorphic features, congenital coronary heart illness, hepatosplenomegaly, irregular platelet aggregation Intrauterine progress retardation, postnatal short stature, disproportionately large head, distinct craniofacial and dental anomalies, distal limb anomalies, particularly brachydactyly and symphalangism. Moderate to extreme intellectual incapacity Cutis laxa in infancy, skinny pores and skin with outstanding veins particularly on the scalp. Although autosomal dominant cutis laxa is taken into account to be a milder type of the disease, systemic involvement can range from mild to extreme, including bronchiectasis and emphysema, hernias, mitral and tricuspid valvular prolapse, pulmonary stenosis, aortic and arterial dilatation and tortuosity, gastrointestinal and urogenital diverticuli [8,13]. In the biggest collection reported to date, 35% had lung and 57% aortic involvement [12]. Herniae, diverticula, arterial tortuosity and aneurysms, severe pulmonary emphysema and cor pulmonale and developmental delay are variable however essential issues. Lysyl oxidase is a serious copperdependent enzyme, and its exercise is markedly decreased in some sufferers [20], resulting in faulty collagen and elastin crosslinks. Clinical manifestations embrace the event of bladder diverticula throughout childhood, inguinal herniae, delicate laxity of the skin and skeletal defects corresponding to short humeri and clavicles. Acquired cutis laxa (see Chapter 96) might rarely develop at any age but often follows an inflammatory process or an related disorder. It is attributable to deletion of the Williams�Beuren syndrome chromosome region (chromosome 7), containing a number of genes [3]. Synonyms and inclusions � Williams syndrome Pfeiffer Saethre� Chotzen Beare� Stevenson 8p11. There could also be circumscribed folds of lax pores and skin in neurofibromatosis, and unfastened folded skin can also happen in leprechaunism, Patterson syndrome and trisomy 18, but these circumstances are distinguished by their related options. Excessive pores and skin wrinkling of the brow has been reported in Apert syndrome [24], although this and the opposite craniosynostosis syndromes are simply distinguished by their craniofacial dysmorphism and recognized associations (Table 72. Introduction and general description Williams�Beuren syndrome is a rare genetic developmental disorder. Epidemiology Incidence and prevalence Approximately 1/10 000 individuals affected [4]. Disease course and prognosis the prognosis is determined by the subtype of cutis laxa however in general autosomal dominant types tend to have a greater prognosis. Neurological examination and imaging is necessary in autosomal recessive cutis laxa type 2. Management Treatment is proscribed and is directed towards assuaging complications. Botulinum toxin has been used successfully as a much less invasive modality to improve facial defects in one case [26]. Although parenteral copperhistidine therapy has been of profit to patients with Menkes disease, it appears to have less impact on the connective tissue abnormalities [27]. The deletion arises on either the maternally or the paternally inherited chromosome 7 and is sporadic in virtually all circumstances [7]. Complications and comorbidities Endocrine abnormalities happen and embrace variable hypercalcaemia, sometimes with related signs [15], impaired glucose tolerance [16] and subclinical thyroid illness [17]. Clinical features Disease course and prognosis Cardiovascular-associated mortality is 25�100 instances that amongst controls [19]. Presentation the syndrome is characterized by untimely laxity of the skin, congenital coronary heart illness (notably supravalvular aortic stenosis), metabolic abnormalities and dysmorphic facial options, which embody a flat nasal bridge, quick upturned nostril and saggy connective tissue around the eyes [1]. A latest study demonstrated additionally an increased incidence of wrinkles (92%), and irregular scarring (33%) as nicely as abnormal biomechanical properties of the skin [11]. Regular monitoring of calcium levels and investigation of other endocrine associations should be performed. Michelin tyre baby this time period refers to a most probably very heterogeneous group of issues manifesting with circumferential skin folds [1]. This condition has been proven in some instances to result from underlying easy muscle hamartomas [2] or adipose tissue hyperplasia [3]. The phenotype can present spontaneous remission [4] and has been described either in isolation or in association with further anomalies [5]. The dysfunction could be sporadic or inherited as an autosomal dominant or recessive trait [1,6].
Elimite 30 gm buy discount online
Reported cases with blisters and tonofilament clumping are prone to acne help order elimite 30 gm without prescription correspond to an epidermolytic or keratinopathic disorder [1�4] skin care 7 belleville nj cheap elimite 30 gm on line. Pathophysiology Studies of a household from Bothnia in northern Sweden and three English pedigrees confirmed linkage centromeric to the type 2 keratin gene cluster on chromosome 12 [16�18]. Aquaporins are a family of cell membrane proteins that allow the osmotic motion of water across the cell membrane. The causative mutations exert a gainoffunction impact resulting in elevated keratinocyte water uptake quite than transepidermal water loss [19]. Hyperhidrosis is usual, and dermatophyte infections and pitted keratolysis are frequent. Otherwise, histological changes are nonspecific: orthokeratotic hyperkeratosis, hypergranulosis or normogranulosis and reasonable acanthosis are often seen. Mal de Meleda Definition and nomenclature Mal de Meleda is a uncommon autosomal recessive transgredient keratoderma named after the Croatian island of Meleda (Mljet) where it was first identified [1,2]. Almost all instances happen in consanguineous pedigrees and families from Croatia (including Meleda), Algeria, Israel and Tunisia share very few ancestral haplotypes, indicating founder mutations [3,6�8]. Haplotype evaluation in Western European sufferers with the disease confirmed a founder effect for the W15R mutation [9]. The gene is expressed late in epidermal differentiation, within the granular layer, and particularly in association with the acrosyringium [3,11]. The erythematous component usually persists in central palms and soles, usually with hyperhidrotic maceration and malodour. Circumferential hyperkeratosis of the fingers may lead to sclerodactyly and digital constrictions (pseudoainhum) (Table sixty five. Hyperpigmented spots [18], melanoma arising within affected skin [19,20] and Bowen illness of the sole have been reported [21]. Heterozygous female carriers might have a light medical phenotype [22], also pseudodominant inheritance has been reported [23]. A outstanding perivascular lymphohistiocytic infiltrate could additionally be seen, sweat glands may be enlarged. Electron microscopy indicates a less abrupt transition from granular to cornified layers [18]. Excision of keratoses and splitthickness pores and skin graft can be an choice in order to relieve useful impairment. Longterm followup in a single patient demonstrated no recurrence of keratosis on surgically handled areas [30]. Loricrin keratoderma Definition and nomenclature In two related pedigrees, Camisa and Rossana et al. Synonyms and inclusions � Variant Vohwinkel syndrome � Mutilating keratoderma with ichthyosis � Camisa syndrome Investigations In addition to hyperkeratosis, histological options include hypergranulosis and parakeratosis, i. Electron microscopy exhibits dense intranuclear granules in granular cells, and a thin cornified cell envelope within the decrease cornified layers with irregular extracellular lamellae [13]. Several different single nucleotide insertions have been identified in this gene, which uniformly end in frame shift and lead to expression of an abnormal protein with an irregular, arginine wealthy Cterminal peptide containing nuclear recognition indicators [3�11]. Direct or oblique consequences embrace moderate alterations on the cornified envelope, elevated corneocyte fragility and irregular epidermal barrier function with accelerated restore kinetics [13]. Transgenic mice in whom loricrin has been knocked out are largely asymptomatic [14], however mice expressing a pathogenic loricrin mutation confirmed generalized scaling, thickened footpads and a constricting band inflicting autoamputation of the tail [15,16]. Clinical features [4,7�11] Generalized desquamation may be noted at delivery, and collodion infants are reported [6,7,10]. The edges of the keratoderma are diffuse (in contrast to true Vohwinkel syndrome) and cicatricial bands (pseudoainhum) may develop across the digits. Today, several distinct keratoderma with striate and/or focal pattern are recognized; furthermore, a few of them may be related to a syndromic entity. Mechanical stress is essential; ache, hyperhidrosis and delicate hyperkeratosis of the knees have been reported [2]. The presence of different options, particularly woolly hair, must be specifically sought, and the potential for cardiac illness considered. Pathophysiology Initially, autosomal dominant striate keratoderma was mapped to the desmosomal cadherin cluster on 18q12. Punctate palmoplantar keratoderma (a) Definition and nomenclature this autosomal dominant keratoderma is clinically characterized by small rounded papular lesions on the palms and soles that are inclined to coalesce over strain factors [1]. Pathophysiology the disorder has been mapped to two chromosomal regions 15q22 [5�7] and 8q24. Enviromental components and personal skincare regimes could affect the degree of hyperkeratosis [18]. In many families, small and large lesions coexist, including broader focal plantar callosities. Importantly, HowelEvans syndrome should be considered if focal and nummular keratoderma predominate and histology is unspecific (see Tylosis with oesophageal cancer later). Investigations Punctate lesions are orthohyperkeratotic on histology with compact acanthosis and hypergranulosis with a depression within the centre of the lesion, but may present hypogranulosis and (focal) parakeratosis [1]. Literature stories indicate no main helpful effects of keratolytic ointments as nicely as topical retinoids or topical calcipotriol on the keratoses [1,23,24]. Systemic treatment with oral retinoids may yield a small effect, moreover relying on the dosage (0. Receptor tyrosine kinase inhibitors which are concerned in p34 signalling are beneath growth and of potential relevance for future treatment [9,26]. Small even keratotic papules on the palms (a) and confluent hyperkeratosis on the feet of the same affected person (b). Discomfort could be caused by an inclination to catch on clothes and other objects [9]. Differential prognosis Most important, focal keratoderma associated with malignancies similar to breast and colonic adenocarcinoma [19,20] must be differentiated (see HowelEvans syndrome later). In acrokera- Differential diagnosis Darier disease [10,11], epidermodysplasia verruciformis [12], arsenic keratosis [4], a number of filiform verrucae, paraneoplastic follicular hyperkeratosis [13] and/or multiple minute digitate hyperkeratosis (reviewed in [4]) should be differentiated. Investigations Costa [5,6] reported 13 cases with cornified and umbilicated papules distributed alongside the borders of the arms and toes. He noted fragmentation and rarefaction of elastic fibres within the dermis, and launched the time period acrokeratoelastoidosis. Around one third of black individuals but hardly ever white could show hyperkeratotic pits involving the flexural creases of the palms [17,18]. Keratoelastoidosis marginalis [19,20] is an acquired condition also referred to as digital papular calcific elastosis. It presents with degenerative collagenous plaques that are agency, generally concave, forming a linear band principally around the internet of the thumb and index finger at the margin of the volar and dorsal surfaces. The absence of dyskeratosis and vacuolated keratinocytes under the parakeratotic columns differentiate the disease from porokeratosis [3,15]. Autoimmune screen, blood investigations for full blood count, renal/liver function and radiological tests are necessary to exclude an related situation, The key microscopic discovering in acrokeratoelastoidosis is the presence of massive elastosis, whereas no elastic fibre changes are current in focal acral hyperkeratosis [1,13]. Cole disease Definition and nomenclature this genodermatosis features punctuate keratoderma and pigmentary anomaly.
30 gm elimite purchase
These studies highlight the necessity for cautious counselling of patients and their households and continued vigilance acne in children order elimite 30 gm amex. Concomitant morphoea skin care zinc oxide cheap elimite 30 gm mastercard, particularly plaque and nodular types, have been reported in up to 6. Investigations the prognosis of morphoea is largely medical, but a variety of investigations can be useful in guiding management (Table fifty seven. A biopsy can be undertaken if the analysis, the depth of involvement or the degree of exercise are doubtful. An incisional ellipse through the violaceous/inflammatory fringe of a lesion, into the sclerotic centre and extending down into the subcutis, must be taken and the location clearly indicated for the pathologist. If the depth of involvement is in question the biopsy ought to lengthen into the fascia and underlying muscle and referral to a plastics or common surgeon could additionally be required. Are there extracutaneous manifestations (headache, migraine, seizures, arthralgia, myalgia,dyspepsia, Raynaud phenomenon, and so on. Serial ultrasonography can be utilized to consider pores and skin thickness and lack of muscle and fat [316]. Disease exercise may be correlated with echogenicity and tissue blood circulate measured by Doppler [317] and laser Doppler [318,319], ultrasound strategies [317,320] and infrared thermography [321]. A durometer is a handheld instrument that measures the depth of pores and skin indentation after the application of a standardized quantity of drive. Durometry measures pores and skin hardness, a acknowledged surrogate for pores and skin thickness [322], and has been proven to objectively discriminate affected versus unaffected skin on the trunk and limbs in kids with morphoea [323]. Increased ranges of creatine kinase and aldolase have been associated with the development of recent lesions, muscle atrophy and limb shortening, and may thus suggest muscle involvement and presumably disease activity [30]. Frequently related autoimmune diseases such as autoimmune thyroid illness ought to be excluded. Defining end result measures in morphoea has proved troublesome because morbidity is brought on by a mixture of cutaneous and subcutaneous injury in addition to illness activity. Trying to discover correct measures of disease activity has been a particular challenge. It has shown high specificity and sensitivity for assessing disease exercise [317]. The most accurate sonographic signs of activity are elevated subcutaneous echogenicity and increased cutaneous blood move. Scanning laser Doppler imaging had a constructive predictive value of 87% and negative predictive worth of 94% in a small study evaluating its use in assessing activity and predicting progression [319]. Clinical scoring strategies have the advantage of not requiring costly or cumbersome tools. Disease harm was outlined as irreversible or persistent modifications of the lesion because of earlier energetic disease or issues of therapy. The cutaneous manifestations embrace hyper/ hypopigmentation and subcutaneous and dermal atrophy. Management the administration of morphoea has been challenging due to a scarcity of standardized analysis methods and of evidencebased remedies. Linear and generalized types of morphoea can lead to permanent cosmetic and useful impairment. However, therapy of sufferers with important damage however inactive illness exposes them to the potential unwanted effects of the medicines, without providing significant benefit. Treatment choices should be based mostly on the subtype of morphoea, the extent of disease exercise and, to some extent, on the age of the affected person. In follow nevertheless, this has not always been the case and the treatment prescribed may rely more on the specialty of the treating doctor than the affected person or their sort of disease. Thus, from a total of 531 prescriptions for 224 sufferers (95 children), dermatologists prescribed topical corticosteroids in 41%, different topical treatment in 25% and phototherapy in 16% of circumstances, and systemic corticosteroids in 5% and methotrexate in 4% of cases. Rheumatologists, in distinction, prescribed methotrexate in 34% and systemic corticosteroids in 31% of instances, and topical therapies in 10% and phototherapy in solely 2% [34]. In this examine, 68% of patients with linear morphoea obtained topical corticosteroids as the mainstay of remedy in the event that they noticed a dermatologist (4% obtained methotrexate), whereas 39% of linear morphoea sufferers seen by rheumatologists acquired methotrexate and 8% topical corticosteroids [34]. There was a broad range of responses: methotrexate with corticosteroids was essentially the most frequent response (37. The initial evaluation of a affected person with morphoea ought to include an assessment of the sort, extent and activity of illness (see Table 57. Treatment can then be primarily based on the subtype, exercise, extent and depth of illness, and affected person signs [2,25,26]. Case reports and a prospective openlabel research in 13 patients initially suggested a task for topical tacrolimus zero. Two small placebocontrolled trials support the utilization of twice daily topical tacrolimus 0. The latter was additionally discovered to be safe and beneficial when used 3�7 occasions weekly, over a 9 month interval, in 21 grownup and paediatric sufferers in two potential openlabel studies [328,336]. Benefit in potential however uncontrolled studies has been proven for twicedaily application of topical vitamin D analogues (calcipotriol/calcipotriene zero. Early inflammatory and sclerotic lesions seem to reply most favourably, and instances with deep involvement least favourably. Outcome measures recorded in over 90% of cases embody variable combos of medical examination, skin scores, ultrasound measurement of skin thickness, cutometer measurements and pores and skin biopsies. The length of responses is variable and as much as half of the patients could develop recurrence of active morphoea lesions at 2�3 years [351]. Nevertheless, lowdose remedy should still have a role, particularly if mixed with topical modalities similar to vitamin D analogues [339]. In patients with progressive illness regardless of topical brokers and/or phototherapy, and in sufferers with linear, deep or disseminated forms of illness similar to morphoea en coup de sabre, pansclerotic morphoea or eosinophilic fasciitis, systemic therapy is indicated. There is broad agreement that mixtures of pulsed intravenous and/or oral steroids with methotrexate must be used first line. An open examine of 17 sufferers with severe morphoea (linear/ generalized) found that oral corticosteroids (0. Systemic corticosteroids must be thought-about in patients with extreme, lively inflammatory illness and in sufferers with eosinophilic fasciitis who seem particularly steroid responsive [253]. Methotrexate is a cornerstone of morphoea administration [363� 365,366,367,368,369,370). Furthermore, mast cell numbers and levels of tenascin � an extracellular matrix protein beforehand shown to be elevated within the pores and skin and circulation of morphoea sufferers [376,377] � are both lowered in lesional pores and skin after methotrexate remedy [378]. Two early uncontrolled case series (17 sufferers, 9 adults) advised some improvement in pores and skin lesions with methotrexate alone [262,363]. Four retrospective evaluations documented the response to methotrexate alone in fifty two instances, and in combination with corticosteroids in 67 instances [365,366,379,380]. Improvement was described in 79% of instances but was extra variable in those treated with methotrexate alone. In three prospective research, a total of 60 sufferers (15 adults) have been treated with both month-to-month pulsed intravenous (1 g, three days per thirty days, for 6 months in the adults and 30 mg/kg, 3 days per month, for three months in 9 children) or daily oral corticosteroids (2 mg/kg/day (maximum dose 60 mg/day), tapered to zero. A 50% discount in skin scores, corroborated by biopsy and ultrasound measurements, was documented in 13/15 adults after a imply treatment period of 9. This profit was confirmed in a randomized placebocontrolled trial in 70 youngsters with energetic linear, generalized or blended morphoea, evaluating 12 months of oral methotrexate (15 mg/m2/week (maximum dose 20 mg/week), n = 46) with placebo (n = 24) [367].
Purchase 30 gm elimite fast delivery
Several pores and skin biopsies may be required for the affirmation of the prognosis of urticarial vasculitis [5] acne scar treatment buy 30 gm elimite overnight delivery. All sufferers with urticarial vasculitis should undergo a laboratory workup consisting of full blood rely acne in ear purchase elimite 30 gm fast delivery, blood biochemistry and erythrocyte sedimentation rate. Urinalysis and liver operate checks are important in laboratory workup for systemic involvement. In the case of irregular urinalysis, 24h urine protein and creatinine clearance must be checked. Antibody screen in sufferers with urticarial vasculitis ought to embrace antinuclear antibodies, antibodies in opposition to extractable nuclear antigens, rheumatoid factor and circulating immune complexes. For instance, suspicion of pulmonary involvement should set off a workup including chest Xray and lung function testing. Patients with severe urticarial vasculitis current with hypocomplementaemia, systemic involvement or therapy refractory disease. Complications and comorbidities Patients with urticarial vasculitis could current with renal involvement (microscopic haematuria or proteinuria) at disease onset or later within the disease course but it not often progresses to renal failure. Chronic obstructive pulmonary illness is taken into account as a life threatening late complication of urticarial vasculitis [2]. Connective tissue illnesses and haematological malignancies are frequent comorbidities in urticarial vasculitis [6] (see disease associations later). In some sufferers, urticarial vasculitis may be the first presentation of these illnesses whereas in others urticarial vasculitis can current in the context of these diseases. Chronic viral infections (hepatitis B and C) are other important comorbidities in urticarial vasculitis. Management Management of urticarial vasculitis is generally based mostly on case stories, small affected person series and a few openlabel, noncontrolled Table forty four. Patients with normocomplementaemic urticarial vasculitis restricted to the pores and skin tend to have a benign illness with a great prognosis. Conversely, hypocomplementaemic urticarial vasculitis is related to a extra severe course and more frequent systemic involvement [2]. Lesional pores and skin biopsy (diagnostic) Full blood depend Erythrocyte sedimentation price Biochemical profile C3, C4 complement parts (serial testing) Antinuclear antibodies Antiextractable nuclear antigens Hepatitis B and C serology Circulating immune complexes Urinalysis Key references forty four. In unresponsive patients, corticosteroids (prednisolone at doses of forty mg/day or more) can then be considered for shortterm administration [5,6,44]. For severe refractory instances, immunosuppressive brokers (cyclophosphamide, azathioprine, and so on. Other approaches such as intravenous immunoglobulins, methotrexate, intramuscular gold and plasmapheresis have additionally been used [48�51]. Recently, several organic therapies have shown promise for urticarial vasculitis in anecdotal reports or small collection. A case of normocomplementaemic urticarial vasculitis exhibiting a partial response to omalizumab (antiIgE) has just lately been reported [54]. Integration of biological agents into the administration protocol for urticarial vasculitis sooner or later could help overcome the issue of toxicity associated with the usage of conventional remedies for urticarial vasculitis, especially longterm oral corticosteroids. The alternative of remedy should take comorbidities and illness associations into consideration. For instance, patients with urticarial vasculitis with systemic lupus erythematosus might reply to dapsone [16]. Stratification of patients when it comes to systemic involvement and prognosis could facilitate extra targeted and individualized remedy approaches in the future. This could additionally be achievable by collaborative multicentre and multidisciplinary efforts given the rarity and complexity of this disease. From a medical perspective, a consensus on the management of urticarial vasculitis is much wanted and would harmonize the remedy approaches to this uncommon situation. Hypocomplementemic urticarial vasculitis syndrome: an interdisciplinary challenge. Cellular and molecular dynamics in exercise induced urticarial vasculitis lesions. First line therapies n Nonsedating H1 antihistamines Nonsteroidal anti-inflammatory drugs Second line treatments Dapsone Colchicine Hydroxychloroquine Short trials of corticosteroids Third line remedies h Azathioprine Ciclosporin Mycophenolate mofetil Methotrexate Intravenous immunoglobulins Cyclophosphamide Cutaneous medical features might embody urticarial reactions, oedema, erysipelaslike lesions, pustulosis and pyoderma gangrenosum. There is also an rising variety of advanced polygenic inflammatory disorders by which abnormalities of the innate immune system play an necessary position. In these autoinflammatory syndromes which current with an urticarial or maculopapular rash, the primary lesion is a red or pink macule or a slightly raised plaque. However, on biopsy a neutrophilic perivascular and interstitial infiltrate of the dermis is present, without vasculitis or important oedema. For all the genetic autoinflammatory syndromes and associated advanced issues, analysis is suspected on scientific or clinico- pathological grounds and confirmed by more particular analyses. Skin biopsy is nearly always indicated and demonstrates the character of the inflammatory infiltrate. Serum amyloid A ranges are additionally increased and that is predictive of an increased risk of amyloidosis, the major complication of many autoinflammatory syndromes. Genetic diagnosis is often the following step and might be guided by the scientific context. Other investigations will rely upon the specific dysfunction, as for example listening to tests within the cryopyrinopathies. Inflammatory flares can occur in plenty of organs, especially the pores and skin, joints, eyes and serous membranes. An underlying genetic abnormality predisposes the affected person to activation of the innate immune system and an exaggerated inflammatory response to exogenous or endogenous triggers. Their relevance reaches far beyond an understanding of their function just in this group of illnesses. Introduction and basic description the person monogenic autoinflammatory syndromes embody the periodic (hereditary) fever syndromes and a variety of syndromes of which pustular eruptions form a component. The spectrum of autoinflammatory disease is constantly expanding as new entities are being described and/or characterized genetically, typically in only a few people or households. They are normally categorised either by clinical criteria, especially mode of inheritance, main signs and kind of rash, pattern of recurrence and period of the inflammatory flare, or according to the genetic abnormality. The detailed phenotypical evaluation and the pathogenic deciphering of monogenic autoinflammatory syndromes have led to a new classification of inflammatory illnesses in general [3]. Epidemiological and medical options of the main monogenic autoinflammatory syndromes are summarized in Table 45. The highest prevalence charges are found in the Sephardic Jewish, Turkish, Armenian and Arab populations: prevalence charges of 1: 248 to 1: 1000 and provider charges of 1: three to 1: 7 are reported [4].
Baikal Skullcap. Elimite.
- Are there any interactions with medications?
- Dosing considerations for Baikal Skullcap.
- Inflammation of small air passages in the lung (bronchiolitis) and other lung infections; kidney, stomach, and pelvic infections; hayfever; seizures; HIV/AIDS; nervous tension; hemorrhoids; prostate cancer; hepatitis; sores or swelling; osteoarthritis; fever; headache; red eyes; flushed face; psoriasis; and bitter taste in the mouth.
- How does Baikal Skullcap work?
- Are there safety concerns?
- What is Baikal Skullcap?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96869

Discount elimite 30 gm otc
Age Prurigo nodularis impacts patients throughout all ages acne active elimite 30 gm without prescription, together with kids [8] acne in your 30s cheap 30 gm elimite with visa, however is extra frequent in older patients [9]. Sex Prurigo nodularis impacts each males and females, however is extra widespread in girls [10]. Prurigo nodularis is often disseminated bilaterally and symmetrically over the entire pores and skin, but preferentially in the dorsal parts of extremities, d�collet�e, back and buttocks. In many patients, the central again is spared, and this zone is designated as the butterfly sign [23]. One complication that may hinder healing is automatic scratching behaviour that patients might need developed; on this case, they continue to scratch even within the absence of itch sensations. Laboratory � primary (frequent associations reported) Laboratory � advanced (rare associations reported) Skin 83. In transient, therapy must be multimodal including therapy of the underlying illness, use of topical emollients (especially in dry pores and skin, atopic predisposition) and topical substances for shortterm relief of itch. A direct immunofluorescence test must be performed if a patient had reported blisters and/or if erythema or blisters have been found on physical examination (Table 83. In another collection, a large number of comorbidities Otolaryngology Psychosomatics/ psychiatry Neurology Iking et al. The population density of nerve fibres is commonly elevated in lesional skin as demonstrated by S100 staining and mast cell numbers may also be increased. There is also upregulated expression of neuropeptides in lesional skin, including tachykinins [2]. Spongiosis is usually current, and small areas of parakeratosis are occasionally seen. Clinical options Characteristically, the historical past is of repeated bouts of intense itching and scratching, interspersed with itch�free intervals. With time the redness subsides and the affected pores and skin becomes pigmented, thickened and slightly scaly. The ordinary websites are the nape of the neck, the perimeters of the neck, the decrease legs and ankles, the scalp, the upper thighs, the vulva, pubis or scrotum, and the extensor forearms. Management the character of lichen simplex and the want to break the scratching behavior have to be explained. Treatment is aimed at breaking the itch� scratch cycle, and suppression of pruritus, often by topical or intralesional corticosteroids. The software of an occlusive dressing or an occlusive bandage will intensify the impact of topical steroids. For very continual lesions, systemically applied substances to suppress pruritus corresponding to gabapentin could be helpful. Clinical classification of itch: a place paper of the International Forum for the Study of Itch. Quality of life and work productivity impairment amongst psoriasis patients: findings from the National Psoriasis Foundation survey data 2003�2011. Serum autotaxin is elevated in pruritus of cholestasis, but not of other origin, and responds to therapeutic interventions. The particular dermatoses of pregnancy revisited and reclassified: outcomes of a retrospective twocenter research on 505 pregnant sufferers. A Practical Treatise on Disease of the Skin for the Use of Students and Practitioners, eighth edn. Prurigo as a symptom of atopic and nonatopic ailments: Aetiological survey in a consecutive cohort of 108 patients. Gender differences in persistent pruritus: women present completely different morbidity, extra scratch lesions and better burden. Reduced intraepidermal nerve fibre density in lesional and nonlesional prurigo nodularis skin as a potential signal of subclinical cutaneous neuropathy. Genital dysaesthetic syndromes (vulvodynia, vestibulodynia, penoscrotodynia) are also common and are mentioned on this chapter, as nicely as in particular genital chapters. There is a rising physique of proof that these sufferers are best managed in specialist, multidisciplinary clinics. The dysaesthetic syndromes comprise a heterogeneous group of problems during which there are abnormal sensations of the skin, usually in a selected web site. There could additionally be little, or nothing, to see by the use of cutaneous clinical indicators, or there may be proof of excoriation, rubbing or skin traumatization. The issues could also be divided into those with no demonstrable neurological cause and those with a believed or demonstrable neurological trigger (Table eighty four. Synonyms and inclusions � Glossodynia � Stomatodynia � Oropyrosis � Scalded mouth syndrome � Glossopyrosis � Glossalgia Introduction and basic description Burning mouth syndrome is a dysfunction notably encountered in perimenopausal women. Associated diseases Patients often have a history of mental well being issues, most commonly anxiousness and depression [1]. Pathophysiology Burning mouth syndrome is most likely a form of neuropathic ache and a selection of hypotheses have been postulated. It is known that some people have increased numbers of style buds (so referred to as supertasters). Part 7: Psychological, sensory & neurological Clinical features History Patients current with a historical past of a number of months to several years of accelerating oral burning, pain or discomfort. Patients may also describe altered style sensations (dysgeusia) and even adjustments in salivation (most incessantly dryness or xerostomia). The burning ache could additionally be triggered or exacerbated by certain (usually spicy or acidic) foods. However, in most patients, food or drink alleviates the pain, at least, quickly. Some sufferers have a habit of thrusting their tongue against the lower anterior teeth in an try to alleviate symptoms. Although some describe comparatively mild pain, many describe it as essentially the most extreme ache possible. Complications and comorbidities Burning mouth syndrome may be associated with affective illness similar to anxiousness and despair. Spontaneous remission rates of 20% after 7 years or 3% after 5 years have been reported [4]. Differential analysis the differential prognosis is that of natural intraoral disease which ends up in intraoral ache. Symptoms vary in severity all through the day, being worse in path of the top of the postherpetic neuralgia 84. If the seventh cranial nerve is involved the acute phase is termed Ramsay Hunt syndrome. Negative Burning mouth syndrome Reassurance, information Cognitive behaviour therapy Topicals Clonazepam Lidocaine Benzydamine Systemic Alpha lipoic acid Antidepressants Introduction and basic description Postherpetic neuralgia presents with a persistent burning, stabbing or itchy ache with sharp exacerbations and associated sensory adjustments. If involving the trigeminal nerve, the ensuing facial and or eye pain may be extreme.
Buy cheap elimite 30 gm on line
Therefore acne treatment for teens purchase elimite 30 gm otc, amyloidoses affecting the skin should be distinguished based on each medical and biochemical investigations skin care urdu purchase elimite 30 gm with visa. This is much like the findings in systemic amyloidosis with cutaneous involvement where subpapillary layers (stratum reticulare, subcutis), dermal appendages and blood vessels may be concerned [48]. Functional and diseasecausing amyloids Clinical presentation In current years, it has been found that amyloid proteins additionally fulfil various biological tasks [33�36], whereas others result in serious diseases, such as Alzheimer illness [37]. Functional amyloid not only occurs in mammals, but additionally in insects, fungi and micro organism [38]. In Homo sapiens, Pmel17 serves as a structural scaffold for covalent polymerization of molecules throughout melanin assembly; chorion S18/S36 is a structural protein of insect egg shells. Aspergillus fumigatus); and bacteria like Escherichia coli and Salmonella typhimurium want main curlin subunits (csgA) for biofilm formation and host invasion [34,38�43]. Other attention-grabbing functional amyloids are particular adhesins for mobile aggregation in yeast cells [44] or sort I antifreeze protein (icestructuring protein preventing ice growth) in winter flounders [45]. These examples present how nature uses the very steady amyloid structure for proteins concerned in safety from environmental influences. Additionally, it has been discovered that peptide and protein hormones in secretory granules of the mammalian endocrine system are generally saved in an amyloidlike cross sheetrich conformation [46]. Localized cutaneous amyloidoses typically present yellowish or brownish macules, papules or plaques of varying configuration, whereas cutaneous amyloidosis as a result of systemic illness usually initially presents with petechiae, ecchymosis and nonhealing ulcers. These differences in clinical presentation are due to the amyloid deposition, which in localized cutaneous amyloidoses happens kind of exclusively in the papillary dermis, whereas it additionally impacts deeper skin layers in systemic amyloidoses. Amyloid precipitation in blood vessel walls makes vessels fragile, ultimately inflicting intracutaneous micro and macrohaemorrhages. Investigations the most important diagnostic step is a lesional skin biopsy, which ought to be sent for histology, immunohistochemistry and electron microscopy (Box fifty eight. This evaluation ought to include a full historical past and bodily examination along with an electrocardiogram, full blood depend, serum creatinine level, serum liverassociated enzymes levels, serum protein electrophoresis and urine protein electrophoresis. It has additionally been advised that an belly fats biopsy (easy entry for screening) can be performed to rule out systemic illness [47]. Any indication of Basic classification Various classification techniques for amyloidoses affecting the pores and skin have been proposed. Extensive amyloid deposits are visible as lightblue precipitates throughout the papillae. Original magnification 20x (plasticembedded, double staining with toluidin blue and alkaline fuchsin). Another method of detecting specific amyloid subtypes is to use in situ hybridization. In latest years, stomach fat pad fineneedle aspiration biopsy has turn into an essential diagnostic software for diagnosing systemic amyloidosis. Even although only minimal quantities of amyloid are present, a definitive prognosis of systemic amyloidosis could additionally be achieved by scanning subcutaneous tissue samples or biopsies from the rectal submucosa with an electron microscope. Amyloidoses could even be subclassified based on the totally different underlying amyloid protein by electron microscopy after immunogold labelling [55]. Apart from organ biopsies, biopsies taken from the rectal submucosa or In localized cutaneous amyloidoses, amyloid precipitates are limited to the pores and skin, principally to the papillary dermis. Hereditary and nonhereditary forms of localized amyloidoses could be differentiated. Type of amyloidosis (amyloid subtype) Amyloid fibril r precursor Cutaneous findings and common n distribution (less common) b 58. In white people, hereditary cutaneous amyloidoses are very rare and only some familes have been reported [58,59]. Amyloid K is therefore a key feature of localized cutaneous amyloidoses [3,fifty four,seventy nine,80]. In this context, pruritus � which provokes scratching of the pores and skin � is thought to induce additional amyloid K deposition, which in flip leads to intensified pruritus. However, scratching of intact pores and skin could also be the starting point for amyloid K deposition, which is also referred to as friction or brush amyloidosis. Therefore, friction amyloidosis is usually also categorised as a form of secondary cutaneous amyloidosis [83,84]. There are two main components that differentiate nodular amyloidosis from macular and lichenoid amyloidosis. In a genomewide screening of Taiwanese households, evidence for a susceptibility locus on chromosome 5 (5p13. The associated improve in apoptotic keratinocytes leads to the buildup of degenerate keratinous materials, which may trigger pruritus and thereby promote amyloid formation [89]. In 1970, Brownstein and Helwig determined that the chance for this progression was 50% based mostly on a cohort of 10 patients [96]. However, this examine included the unique Brownstein and Helwig study, thus including five sufferers that will have had systemic amyloidosis to start with. Because of this, the true price of development of the 47 instances researched by Northcutt and Vanover is wherever between 5% and 15%. Clinical features Presentation A attribute scientific correlate of cutaneous amyloid precipitates is intense pruritus. In the group of non hereditary systemic amyloidoses with cutaneous involvement, there are major and myeloma or plasmocytomaassociated amyloidoses, secondary amyloidoses associated with irritation or tumours, and haemodialysisassociated forms. Furthermore, amyloidosis cutis dyschromica is a rare variant with only a few cases reported worldwide [100]. The primary medical options are mottled hyper and hypopigmented macules with Cutaneous amyloidoses because of systemic illness 58. As talked about, there are also hereditary systemic amyloidoses and systemic illnesses with secondary cutaneous involvement. In cutaneous involvement, all pores and skin layers in addition to vessel partitions may be affected. For the epidemiology of the completely different forms of cutaneous amyloidoses due to systemic disease, see Table fifty eight. Pathophysiology Most nonhereditary main systemic amyloidoses are attributable to monoclonal plasma cell proliferation. The underlying illnesses comprise entities such as a quantity of myeloma, Waldenstr�m illness, Bence Jones plasmocytoma, heavy chain illness, malignant lymphomas and others. The common feature of those ailments is the production of monoclonal immunoglobulins [47]. Among the suspected causes for dermatorrhagia are that factor X is decreased by binding to amyloid fibrils, and that amyloid deposits in the blood vessel partitions increase vessel wall fragility. Amyloid precipitation inside the oral cavity mucosa might current as papules in a local deposition. Interestingly, experimental cutaneous amyloidosis due to Leishmania infection has been described [111]. Another variant of secondary systemic amyloidosis with cutaneous involvement occurs in haemodialysis patients with 2microglobulin (sheet structure) because the amyloid precursor (Table 58. Clinical options the scientific presentation of cutaneous involvement in systemic amyloidoses is often characterized by petechiae, haemorrhages, ecchymosis and pruritus.

Order 30 gm elimite with mastercard
Patients affected by these types of undernutrition may be identified on the idea of their delayed or decelerated progress parameters skin care products online elimite 30 gm cheap without prescription, clinical options and underlying aetiology acne active discount elimite 30 gm without a prescription. Kwashiorkor refers to the illness state that arises from the prolonged disproportionate consumption of carbohydrate (energy or calories) in extra of different macronutrients, specifically protein. Marasmus, in contrast, refers to the illness state resulting from globally decreased consumption of all macronutrients, together with carbohydrate, protein and fats. Forms of severe malnutrition could be categorised on the basis of specific growth parameters. These have included weight below median weightforage [3], ratio of mid�upper arm circumference to head circumference [4] or abnormally low zscores (standard deviations) below standardized development parameters corresponding to weightforage, heightforage or body mass index [5�7]. A negative number would indicate the variety of normal deviations beneath that median reference. For kids 6�60 months of age, average malnutrition was outlined as having a zscore for weightforheight between �3 and �2 and severe malnutrition noted when the weightforheight zscore was <�3 [8]. In 2005, having a mid�upper arm circumference of <115 mm was recommended as an extra screening measure of undernutrition [9]. These classification schemes have been generally used in characterizing adults with undernutrition, and typically describe three broad categories: malnutrition because of decreased nutrient intake (starvation); malnutrition resulting from acute stress (infectious disease or trauma); and malnutrition secondary to continual disease with related endorgan dysfunction, malignancy or rheumatological disease. These final two forms describe disease states associated to elevated physiological needs outpacing what may otherwise be thought-about normal nutrient intake [10,11]. The prevalence of protein�energy malnutrition or losing syndrome among outpatient adult dialysis sufferers additionally falls into this range [19]. The prevalence of malnutrition generally tends to be larger among adults with increasing age [20]. Age Malnutrition is often a disease seen amongst infants and younger children, and is conventionally evaluated in these beneath 5 years of age. However, protein�energy malnutrition has been properly documented amongst adults, notably those that are aged, debilitated with persistent sickness, or hospitalized [21]. Sex Gender variations in undernutrition are largely determined by regional and cultural differences. It has been hypothesized that scarce resources could additionally be preferentially provided to male children than to feminine youngsters leading to a better prevalence for undernourished girls, significantly among those with more severe malnutrition. No gender variations were noted in a study of hospitalized sufferers in Guangzhou, China [30]. Part 5: Metabolic & NutritioNal Ethnicity the role of ethnicity is confounded by socioeconomic components. For those with severe losing, 71% lived in Asia and 8% in Africa whereas of these with milder losing, 69% were in Asia and 28% in Africa. As a end result, micronutrient deficiencies may also be current, including but not restricted to zinc [32,33], selenium and copper [34], ascorbic acid [35], biotin [36,37], iron [38] and folate [39]. Incidence and prevalence Over the past decade, there was a big improvement in the worldwide burden and prevalence of undernutrition for children beneath 5 years of age. By 2012, the variety of underweight and stunted kids had decreased to ninety nine million and 162 million, respectively [12]. While there has been some variability within the definition of malnutrition in revealed studies, the prevalence of protein�energy malnutrition amongst hospitalized children has various widely and has been estimated at between 4. Prevalence charges of Pathophysiology In kwashiorkor, excess carbohydrate without important protein consumption suppresses insulin production, resulting within the inhibition of protein synthesis [40,41]. Patients with this condition manifest clinically with signs of hypoproteinaemia, including oedema and fatty infiltration of the liver as a outcome of deficiencies in lipoproteins. Those chronically affected also reveal immune suppression as a outcome of decreased production of immune proteins corresponding to immunoglobulins and are therefore in danger for opportunistic infections. Growth retardation is famous but sometimes falls between 60 and 80% of ideal physique weight. Patients with persistent ailments may have physiological wants that exceed obtainable dietary intake. Predisposing components In paediatric protein�energy malnutrition, factors similar to political instability and war, pure disasters, residence in inhospitable environments and economically disenfranchised populations result in suboptimal and imbalanced diets which can be largely deficient in protein and mostly reliant on carbohydrates such as corn, rice or beans. Protein�energy malnutrition is considerably less frequent in industrialized nations. In this setting, this form of malnutrition has been identified in a variety of prototypical conditions. Altered nutrient intake may end up from perceived (real or imagined) meals allergy symptoms. Several circumstances of protein�energy malnutrition have been documented from dad and mom switching from milkbased formulation or milk itself to rice beverages due to perceived dietary milk allergies [43�48]. Hypermetabolic (catabolic) states associated with inflammatory or infectious illnesses can outcome in significantly elevated physiological needs for protein resulting in protein�energy malnutrition. Decreased absorption of protein from gastrointestinal problems such as inflammatory bowel disease [52], proteinlosing enteropathies (seen in association with systemic lupus erythematosus) [53], cystic fibrosis [54] or following bariatric surgery [55]. Finally, decreased synthesis of protein as a result of an underlying metabolic dysfunction has also been suspected as a contributing issue to protein�energy malnutrition in single case reviews of glutaric acidaemia [56] and Hartnup illness [57]. Patients with protein�energy malnutrition are immune suppressed, so special consideration ought to be paid to the potential of comorbid opportunistic infections. Presentation Children with protein�energy malnutrition show signs of failing to thrive. Their development velocity is decreased, and weight and ultimately size percentiles fall. Those with marasmus are inclined to show lack of subcutaneous fat which may end in a wizened or prematurely aged appearance when the buccal fat pads are affected. Abdominal distention is a feature observed in children and has been attributed to hepatomegaly from increased fatty deposition due to lack of accessible apolipoproteins. Over time, more outlined plaques could come up that might be extra concentrated in areas of friction such because the intertriginous areas. The hair usually Pathology Histologically, skin biopsies from those with protein�energy malnutrition show psoriasiform hyperplasia, hyperkeratosis and epidermal pigmentation; atrophy or rete flattening may be current. Causative organisms Evidence from mouse models and one examine in Malawi means that alterations in the gastrointestinal microbiome may additionally serve as a predisposing factor to protein�energy malnutrition [59]. Environmental elements Environmental components could contribute to protein�energy malnutrition by creating a lack of access to needed vitamins. These might embody political or financial instability, war, famine, continual disease and pure disasters. Note the loss of subcutaneous tissue, progress stunting, dyspigmentation and desquamative changes. The reddening of the hair is related to durations of protein�energy malnutrition. Inflammatory changes of the mucous membranes � xerophthalmia, cheilitis, stomatitis and vulvovaginitis � may come up in association with other comorbid vitamin and mineral micronutrient deficiencies [60]. Those with marasmic kwashiorkor sometimes have oedema and show overlapping options of marasmus and kwashiorkor. Differential analysis There are several important concerns in the differential diagnosis of protein�energy malnutrition (Table 63.
Discount elimite 30 gm overnight delivery
These are examples of the place the pathology of each the obesity and skin phenotype are well understood [1 acne treatment for men cheap 30 gm elimite with amex,2] skin care help elimite 30 gm discount online. Prader�Willi syndrome Imprinting is the method by which genetic alleles responsible for a phenotype are inherited from one father or mother only. Genetic associations with lipoma Lipomas, benign tumours of adipocytes, are mentioned in detail in Chapter 137. More recently, mutations in the identical gene have been related to extra restricted phenotypes, including macrodactyly and facial lipomatosis [4,5]. The arrows show a C202T mutation resulting in a untimely stop throughout the protein just upstream of the melanocytestimulating hormone peptide. A8344G mutation) Unknown Gene function Unknown Clinical abstract Encapsulated nonpainful lipomas on the trunk and extremities. Twenty years of polymers: a private perspective on alpha1 antitrypsin deficiency. NakajoNishimura syndrome: an autoinflammatory disorder exhibiting perniolike rashes and progressive partial lipodystrophy. Thymic features and gene expression profile distinct doublenegative cells from single positive cells in the autoimmune lymphoproliferative syndrome. There is one household described with short stature and lipoedema affecting 4 generations. With this new understanding has come the opportunity to reclassify the wellrecognized phenotypes into less complicated and probably extra clinically relevant groups. This new classification comes with the caveat that research on this subject is still revealing findings at a startling fee, and the proposed classification is more doubtless to need to accommodate further adjustments sooner or later. This is the dermatological phenotypic correlate of genotypic mosaicism, defined as the presence of two or more genetically distinct populations of cells inside an individual derived from a single zygote. The phenotype will largely be dictated by the traditional operate of the gene during growth, and the specific mutation that occurs. In congenital naevi there seems to be beautiful speci ficity of genotype�phenotype association, all the method down to codon and even base pair level. The extent and/or number of lesions is a vital (although not absolute) guide to the potential for nondermatological associations. More than one distinct naevus in the same indi vidual suggests a single somatic event affecting a precursor cell earlier than full commitment to one area of the skin, which might due to this fact doubtlessly have led to nondermatological features as nicely; nonetheless, this can be a guideline solely. The cell type affected by the mutation is, of course, central to the clinical phenotype, both in look and distribution. This phenomenon is but to be defined, but must ultimately be associated to the interplay between the 2 genetic populations during devel opment. These can each be divided into: � Cutaneous involvement solely � single or a number of lesions. Histological classification Within congenital epidermal naevi there are subclassifications based mostly on the predominant cell kind seen on histology: � Keratinocytic naevi. Within congenital pigment cell naevi there are subclassifications based mostly on the histology of the pigment cells: � Melanocytic naevus. Within congenital connective tissue naevi there are subclassifi cations primarily based on the predominant cell sort seen on histology: � Collagen naevi. Where this systematic classification leads to a clear prognosis of a clinically welldefined syndrome (such as Proteus syndrome in this case), then the eponymous name must be used if most well-liked. This systematic classification permits for the outline of latest syndromes, for the differentiation of identical medical phenotypes attributable to totally different genetic abnormalities, for the correct description of restricted versions of identified syndromes, and for the approximation of beforehand distinct diagnoses such as Schimmelpenning syndrome and phakomatosis pigmentokeratotica. A clinical diagnosis could due to this fact be initially of a congenital epi dermal naevus syndrome, which with histological and genetic Congenital epidermal naevi seventy five. Data are also missing for whether there are differences between the sexes and ethnic groups. Introduction and general description Congenital epidermal naevi is a descriptive time period for congenital hamartomas of epidermal buildings. This encompasses a extensive range of scientific and histological phenotypes, which can occur as isolated cutaneous lesions or in affiliation with extracutane ous options as a half of various syndromes. In addition, followup of a newborn or younger youngster is recommended for all but small single lesions, as the full phenotype is probably not apparent for a number of years. A very wide range of extracutaneous associations has been reported in the literature, with important overlap between traditionally distinct diagnoses, but with many circumstances appearing to be unique. The significance of correct analysis of the epi dermolytic subtype � which although often suspected clinically should be checked histopathologically � is in the identified associa tion of these naevi with gonadal mosaicism [11], and the flexibility of these mutations to be supported in a germline heterozygous state (see below). Mosaic mutations can solely be passed on if they affect the gam etes of the individual, and are additionally suitable with life as a germ line heterozygous trait in the affected offspring. An essential side of histopathological examination of keratinocytic naevi is the presence or absence of epidermolysis, as when current this distinguishes epidermolytic naevi. Clinical options History Congenital epidermal naevi are by definition current at birth or become apparent within the first years of life. The phenotype can develop in extent over the primary years, and infrequently turn into more pro nounced with age, notably with rising hyperkeratosis in keratinocytic naevi. Keratinocytic naevi range from pale brown and practically macular with a gentle velvety feel, to brown or pink, verrucous or hyperkera totic, and may have a prominent inflammatory element. It is characterized clinically by inflamed and hyper keratotic skin that can be pruritic. Follicular naevi (naevus comedonicus or pimples naevus) are very attribute in look, pores and skin coloured with a excessive density of multiple, comedolike lesions. The number of skin lesions correlates to some extent with the severity of the phenotype in an affected indi vidual [25], and might progress over time [26]. Proteus syndrome as outlined by the diagnostic criteria is attributable to somatic mosaicism Congenital epidermal naevi 75. This newly delineated familial or sporadic syn drome is characterised by multiple, round/ovoid, papular, white/ yellowish, keratotic lesions of 0. Many of the cases described have had gentle neurodevelopmental delay and/or epilepsy [33]. The inheritance is Xlinked dominant, and the condition is almost universally lethal in males. This is the rare affiliation of seg psychological basaloid follicular hamartomas with extracutaneous abnormalities, most notably within the dental, osseous and neurologi cal techniques [39,40]. Naevus come donicus has been described in association with cataracts, skeletal abnormalities and neurological abnormalities [41�43]. This acronym is the description of associ ated congenital lipomatous overgrowth, vascular malformation, epidermal naevi (keratinocytic type) and skeletal abnormalities [29]. This overgrowth syndrome can be related to neu rological abnormalities [30]. This situation requires phosphate alternative, and specialist endocrinology management.