

Deltasone dosages: 40 mg, 20 mg, 10 mg, 5 mg
Deltasone packs: 30 pills, 60 pills, 90 pills, 120 pills, 180 pills, 270 pills, 360 pills

40 mg deltasone best
By bacteria-stasis syndromes (blind loops allergy medicine drowsy 40 mg deltasone with visa, pouches of diverticulosis allergy treatment machine buy deltasone 40 mg lowest price, strictures, fistulas, anastomosis), impaired bowel motility (scleroderma), hypogammaglobulinemia 2. Abnormal mucosal architecture/function-tropical/nontropical sprue, Crohn illness, tuberculous ileitis, amyloidosis C. These lesions unfold all through the size of the wire and ultimately contain spinothalamic and spinocerebellar tracts. There can be degeneration of the dorsal root ganglia, celiac ganglia, the Meissner plexus, and the Auerbach plexus. Nearly one-half had abnormal evoked potential (using the oddball auditory paradigm), which revealed P300 latency that was reversible in 3 months of cobalamin alternative; in one-fifth P300 was unrecordable. Objective checks to document cobalamin neuropathy102 embrace nerve conduction research and motor- and sensoryevoked potentials,103,104 visual pathway abnormalities,a hundred and five and magnetic resonance imaging that shows T2 hyperintensity and atrophy. Among one other cohort of sufferers with cobalamin neuropathy from the United States, 65% had mild, about 25% had moderate, and about 10% had severe neurologic deficits. Physiologic-pregnancy and lactation, prematurity, hyperemesis gravidarum, infancy 2. Intrinsic hematologic ailments involving hemolysis with compensatory erythropoiesis, abnormal hematopoiesis, or bone marrow infiltration with malignant illness b. With mucosal abnormalities-tropical and nontropical sprue, regional enteritis Defective folate transport throughout the choroid plexus into the cerebrospinal fluid-cerebral folate deficiency (mutation or autoantibodies to folate receptors) (rare) Inadequate mobile utilization A. Classic displays of dietary cobalamin deficiency in developing countries are sometimes accompanied by iron deficiency, and amongst malnourished populations, many will also have folate deficiency. Among vegetarians in growing nations, cases with dietary cobalamin deficiency may present within the second and third many years with pancytopenia, delicate hepatosplenomegaly, fever, and occasionally thrombocytopenic bleeding. Because neuropsychiatric shows could dominate the scientific image and the affected person may not have anemia, cautious review of the peripheral smear may reveal macrocytosis. Cobalamin deficiency negatively affects bone development and maintenance,106 which explains the lowered bone mineral density in these with cobalamin deficiency107,108 together with pernicious anemia. Continued unfavorable cobalamin balance leads to an absolute decrease in serum cobalamin degree. The cross section of the spinal cord stained with Luxol blue exhibits demyelination of the dorsal columns (a) and early demyelination of the lateral columns (b). Chapter39 MegaloblasticAnemias ModifiedTherapeuticTrials the standard therapeutic trial utilizing physiologic doses of nutritional vitamins (100 �g of folate or 1 �g of cobalamin given every day while monitoring the reticulocyte response)15 has given method to a modified therapeutic trial. This may be demonstrated by lack of response to full alternative doses of both nutritional vitamins (1 mg of folic acid orally for 10 days and 1 mg of cobalamin intramuscularly or subcutaneously daily for 10 days). Clinical situations in which such trials may be applicable (after drawing blood for serum cobalamin and folate levels) are as follows: 1. The abnormally excessive levels of metabolites return to normal solely when the affected person receives substitute with the appropriate (deficient) vitamin. Conversely, remedy with folate ends in a decrease within the isolated homocysteine degree if folate deficiency is present. This can, nevertheless, be extra vexing within the case of diagnosis of early cobalamin deficiency when symptoms are subtle, nonspecific, or not but fully manifest. However, in elderly patients with anemia, there may be a number of other causes for anemia, and relying on the inhabitants studied, cobalamin deficiency may be only one amongst several possible causes within the differential analysis. This is when a modified therapeutic trial-in which a affected person is handled with full doses of each cobalamin and folate for 10 days-and the dearth of goal response to cobalamin would effectively rule out cobalamin deficiency as a trigger (see field on Modified Therapeutic Trials). Alternatively, attribution of the purpose for anemia to cobalamin deficiency could be moderately confirmed retrospectively if there was evidence for decision of anemia following such therapy with cobalamin. Folate deficiency also ends in elevated ranges of homocysteine due to decreased activity of the methionine synthase�catalyzed reaction. Serum homocysteine concentrations are elevated in each cobalamin deficiency (median values of 70 �M) and folate deficiency (median values of fifty �M). SerumCobalaminLevels For probably the most part, a low serum cobalamin stage is an established biochemical indicator of cobalamin deficiency. In basic, in sufferers with medical cobalamin deficiency and megaloblastic anemia or neurologic disease in preserving with cobalamin deficiency, the sensitivity of cobalamin concentration lower than 200 pg/mL (or less than 148 pmol/L) exceeds 95%116 when the pretest chance is excessive. The serum folate stage is very delicate to folate intake, and a single hospital meal may normalize it in a patient with true folate deficiency. It is important to contemplate the medical image, peripheral smear, and bone marrow morphology and likewise to rule out underlying cobalamin deficiency. Thus a serum cobalamin focus is less than 300 pg/mL in 99% of patients with clinical hematologic or neurologic manifestations of cobalamin deficiency,22 and a cobalamin level of greater than 300 pg/mL predicts folate deficiency or another hematologic or neurologic illness (see Table 39. Among 173 unambiguously cobalamin-deficient patients22 about 5% had regular cobalamin levels. If the serum cobalamin take a look at is broadly used as a screening check with out scientific context, by virtue of the way normalcy is defined, 2. Cobalamin deficiency can falsely raise serum folate by 20% to 30% by way of methyl-folate trapping. This will critically underestimate the prevalence of an associated folate deficiency among populations (mostly in developing countries, worldwide) where the dietary intake of each nutritional vitamins is persistently low. Microbiologic assays for folate, which measure all biologically active forms equally, have been replaced in the West by competitive folate-binding protein assays (from varied industrial sources) which are indirect immunoassays, which rely on chemiluminescence methods. These exams are infamous for appreciable lack of settlement with one another (see box on Diagnosing Folate Deficiency). Alignment with a new higher-order precision isotope-dilution liquid chromatography�tandem mass spectrometry assay, which demonstrates wonderful settlement with the normal Lactobacillus casei method,a hundred and twenty will enable higher standardization of the current aggressive folate-binding protein assays. First, the serum folate degree may be artificially raised in a patient with both pure cobalamin deficiency or combined cobalamin- and folate- deficiency (Table 39. This is because cessation of the cobalamin-dependent methionine synthase response leads to a failure in utilization of intracellular folate for one-carbon metabolism. In these circumstances, serum ranges of metabolites can help within the prognosis of vitamin deficiency. Diagnostic algorithms consistently stress the value of medical information to enhance the pretest likelihood of serum cobalamin and serum folate checks. In combined cobalamin-plus-folate deficiency, both nutritional vitamins can be wanted to restore baseline values, notably of homocysteine. Second, such sufferers (with mixed dietary folate and cobalamin deficiency) usually reside in malarious areas where there may be ongoing hemolysis from malaria per se in addition to intrinsic hemolysis from associated hemoglobinopathies which would possibly be widespread in these areas. In a patient with malaria throughout hemolysis of Plasmodium falciparum contaminated erythroid precursors, reticulocytes, and mature erythrocytes, there shall be substantial launch of the 30-fold more folate-rich intraerythrocyte contents into serum, thereby artificially elevating the baseline serum folate stage. Moreover, red cells normally contain substantial quantities of assorted types of folate, i. In this scientific context, the present assays for serum folate (which are primarily designed to measure physiologic serum 5-methyltetrahydrofolate monoglutamate) may not consistently discriminate amongst these types of folates. This predictable masking of tissue folate depletion argues against the use of serum tests for folate deficiency on this medical setting, where assessing the consumption of folate-rich foods in the diet is a better method to assess folate status. The information introduced on this desk has been synthesized from several sources (see textual content and references).
Diseases
- Balo disease
- Alveolar echinococcosis
- Multiple system atrophy
- Neuropathy, hereditary sensory, type II
- Ventriculo-arterial discordance, isolated
- Cleft palate cardiac defect ectrodactyly
- Chromosome 9, monosomy 9p
- Merlob Grunebaum Reisner syndrome
- Homologous wasting disease
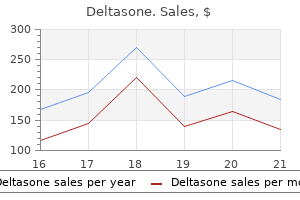
Order deltasone 5 mg line
Ladis V allergy testing taunton deltasone 10 mg order with visa, Theodorides C allergy shots effective for cat allergies deltasone 40 mg generic fast delivery, Palamidou F, et al: Glucose disturbances and regulation with glibenclamide in thalassemia. Masala A, Meloni T, Gallisai D, et al: Endocrine functioning in multitransfused prepubertal sufferers with homozygous beta-thalassemia. Zervas A, Katopodi A, Protonotariou A, et al: Assessment of thyroid operate in 200 sufferers with beta-thalassemia major. De Virgiliis S, Cornacchia G, Sanna G, et al: Chronic liver disease in transfusion-dependent thalassemia: liver iron quantitation and distribution. Di Marco V, Lo Iacono O, Almasio P, et al: Long-term efficacy of alpha-interferon in beta-thalassemics with persistent hepatitis C. Harmatz P, Olivieri N, Kwiatkowski J, et al: Safety and efficacy of peginterferon Alfa-2a and Ribavirin for hepatitis C in thalassemia. Voskaridou E, Terpos E: New insights into the pathophysiology and administration of osteoporosis in sufferers with beta thalassaemia. Terpos E, Voskaridou E: Treatment options for thalassemia sufferers with osteoporosis. Giardina P, Schneider R, Lesser M, et al: Abnormal bone metabolism in thalassemia. In Ando S, Brancati C, editors: Endocrine disorders in thalassemia, Berlin, Heidelberg, 1995, Springer Verlag, p 39. Laor E, Garfunkel A, Koyoumdjisky-Kaye E: Skeletal and dental retardation in beta-thalassemia main. Aydinok Y, Darcan S, Polat A, et al: Endocrine complications in sufferers with beta-thalassemia major. Borgna-Pignatti C, De Stefano P, Zonta L, et al: Growth and sexual maturation in thalassemia main. Scacchi M, Danesi L, De Martin M, et al: Treatment with biosynthetic progress hormone of quick thalassaemic sufferers with impaired progress hormone secretion. Theodoridis C, Ladis V, Papatheodorou A, et al: Growth and management of quick stature in thalassaemia major. Katzos G, Papakostantinou-Athanasiadou E, Athanasiou-Metaxa M, et al: Growth hormone therapy briefly children with betathalassemia major. Eleftheriou P, Tanner M, Pennell D, et al: Response of myocardial T2* to oral deferasirox monotherapy for 1 Year in 29 Patients with transfusion-dependent anaemias; A subgroup analysis. Mariotti E, Angelucci E, Agostini A, et al: Evaluation of cardiac status in iron-loaded thalassaemia sufferers following bone marrow transplantation: enchancment in cardiac function throughout discount in physique iron burden. Perrimond H, Michel G, Orsini A, et al: First report of a cardiac transplantation in a patient with thalassaemia major. Wasi P: Streptococcal an infection leading to cardiac and renal involvement in thalassaemia. Eldor A, Maclouf J, Lellouche F, et al: A continual hypercoagulable state and life-long platelet activation in beta thalassemia major. Rostagno C, Prisco D, Abbate R, et al: Pulmonary hypertension related to long-standing thrombocytosis. Fucharoen S, Youngchaiyud P, Wasi P: Hypoxaemia and the effect of aspirin in thalassaemia. Laopodis V, Kritikos E, Rizzoti L, et al: Laparoscopic splenectomy in beta-thalassemia main patients. Pinca A, Di Palma A, Soriani S, et al: Effectiveness of partial splenic embolization as treatment for hypersplenism in thalassaemia main: a 7-year observe up. Borgna-Pignatti C, De Stefano P, Barone F, et al: Penicillin compliance in splenectomized thalassemics. Angelucci E, Baronciani D: Allogeneic stem cell transplantation for thalassemia major. Lucarelli G, Galimberti M, Giardini C, et al: Bone marrow transplantation in thalassemia. Sodani P, Gaziev D, Polchi P, et al: New method for bone marrow transplantation in patients with class three thalassemia aged youthful than 17 years. Gaziev J, Sodani P, Polchi P, et al: Bone marrow transplantation in adults with thalassemia: Treatment and long-term follow-up. Gaziev D, Polchi P, Galimberti M, et al: Graft-versus-host illness after bone marrow transplantation for thalassemia: an evaluation of incidence and risk elements. Gaziev J, Marziali M, Isgr� A, et al: Bone marrow transplantation for thalassemia from alternative associated donors: improved outcomes with a new strategy. Galanello R, Sanna S, Perseu L, et al: Amelioration of Sardinian beta0 thalassemia by genetic modifiers. Nuinoon M, Makarasara W, Mushiroda T, et al: A genome-wide association recognized the widespread genetic variants affect disease severity in beta0-thalassemia/hemoglobin E. Stamatoyannopoulos G, Veith R, al-Khatti A, et al: Induction of fetal hemoglobin by cell-cycle-specific medication and recombinant erythropoietin. Fucharoen S, Siritanaratkul N, Winichagoon P, et al: Hydroxyurea will increase hemoglobin F levels and improves the effectiveness of erythropoiesis in beta-thalassemia/hemoglobin E illness. Voskaridou E, Kalotychou V, Loukopoulos D: Clinical and laboratory effects of long-term administration of hydroxyurea to sufferers with sickle-cell/beta-thalassaemia. Loukopoulos D, Voskaridou E, Stamoulakatou A, et al: Hydroxyurea therapy in thalassemia. Hoppe C, Vichinsky E, Lewis B, et al: Hydroxyurea and sodium phenylbutyrate remedy in thalassemia intermedia. Nisli G, Kavakli K, Aydinok Y, et al: Recombinant erythropoietin trial in youngsters with transfusion-dependent homozygous beta-thalassemia. Bourantas K, Economou G, Georgiou J: Administration of excessive doses of recombinant human erythropoietin to sufferers with beta-thalassemia intermedia: a preliminary trial. DeSimone J, Koshy M, Dorn L, et al: Maintenance of elevated fetal hemoglobin levels by decitabine throughout dose interval therapy of sickle cell anemia. May C, Rivella S, Callegari J, et al: Therapeutic haemoglobin synthesis in beta-thalassaemic mice expressing lentivirus-encoded human betaglobin. Rivella S, May C, Chadburn A, et al: A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene switch. May C, Rivella S, Chadburn A, et al: Successful therapy of murine beta-thalassemia intermedia by transfer of the human beta-globin gene. Lacerra G, Sierakowska H, Carestia C, et al: Restoration of hemoglobin A synthesis in erythroid cells from peripheral blood of thalassemic sufferers. Baum C, Dullmann J, Li Z, et al: Side effects of retroviral gene switch into hematopoietic stem cells. Breda L, Gambari R, Rivella S: Gene therapy in thalassemia and hemoglobinopathies. Takahashi K, Yamanaka S: Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Okita K, Ichisaka T, Yamanaka S: Generation of germline-competent induced pluripotent stem cells. Aksoy M, Bermek E, Almis G, et al: beta-Thalassemia intermedia homozygous for regular hemoglobin A2 beta-thalassemia.
Deltasone 10 mg low price
Establishing that the affected person does have megaloblastosis is allergy jobs acaai discount deltasone 10 mg without prescription, in principle allergy shots maintenance deltasone 20 mg order with visa, straightforward. If the patient is moderately symptomatic (but not in coronary heart failure), the robust chance of a dramatic response (in the sense of wellbeing and relief of sore tongue) within 2 to 3 days even before hematologic improvement argues against quick blood transfusion. If the patient is properly compensated and within the outpatient setting, the physician has time to develop an orderly sequence of diagnostic tests. If making the prognosis is urgent, a cheap test is the bone marrow aspirate; outcomes indicating megaloblastosis (or not) may be available inside an hour. These checks are notably useful if the affected person has pure neurologic disease or if there are associated conditions such as iron deficiency or thalassemia that may mask megaloblastosis. When the megaloblastic state is established, attempt to determine the underlying mechanism of cobalamin or folate deficiency. The reason for folate deficiency is often sorted out by this time from the history, physical examination, and the scientific setting. A minimalist approach is to work around the nonavailability of the Schilling take a look at and use a basic scientific method to rule out potential differential diagnoses of cobalamin deficiency (see box on Etiopathophysiologic Classification of Cobalamin Deficiency). For instance, most circumstances predisposing to cobalamin deficiency ought to be clinically manifest by the time cobalamin deficiency is obvious. It ought to due to this fact be possible to identify several circumstances through a detailed dietary historical past or previous medical history, travel history, and drug historical past to counsel a dietary cause, esophagogastroduodenal illness, pancreatic insufficiency, impaired bowel motility, or other autoimmune diseases. The historical past and bodily examination might present additional leads and suggest additional focused laboratory testing for rare situations (stool for ova, anti�tissue transglutaminase antibodies, lipase, gastrin, intestinal biopsy, or radiographic contrast studies for stasis, strictures, fistulas). With no further leads, and subsequently by default, one can assume that the prognosis is both pernicious anemia or food-cobalamin malabsorption, which are both treated with comparable substitute doses of cobalamin. Thus the historical past, physical findings, and centered laboratory checks with careful scientific follow-up can potentially determine the cause of the vast majority of cases of cobalamin deficiency and bypass the need for a Schilling check. However, if malabsorption of food-bound cobalamin is suspected (especially in the elderly with achlorhydria), higher doses of every day oral cobalamin (equal to or larger than one thousand �g/day) is required. More than 98% of all the cobalamin in feces is in the type of cobalamin analogues, and about 80% of the ingested cobalamin is transformed to analogues by microorganisms within the intestine. It is simply too costly for standard repletion in folate-deficient states in adults. ResponsetoReplenishment the response of the affected person to applicable replacement is reversion of megaloblastic hematopoiesis to normal hematopoiesis inside the first 12 hours; by forty eight hours, normal hematopoiesis is reestablished, and the only proof for a previous megaloblastic state may be the persistence of a few giant metamyelocytes. Because megaloblastosis attributable to cobalamin or folate deficiency could be reversed in 24 hours by administration of folate. Clinically the primary 36 to forty eight hours are often highlighted by the awakening of an occasional semistuporous particular person whose "chief complaint" is amazement at the remarkably improved sense of well-being experienced, with increased alertness and urge for food and decreased soreness of the tongue. This might precipitate an assault of gout if the patient has a "gouty predisposition. During this process, there may be a transient left shift to embrace myeloid precursors. In response to cobalamin, progression of neurologic harm and dysfunction is inhibited. In common, the degree of useful restoration is inversely associated to the extent of disease and length of signs and signs. As a tough estimate, signs and symptoms that have been has been extended, anticipate related cobalamin deficiency to ensue (special emphasis must be given to figuring out subtle manifestations of neurologic disease). An applicable regimen for situations in which cobalamin replenishment can appropriate cellular cobalamin deficiency (but not correct the underlying problem that led to the deficiency, corresponding to pernicious anemia) is 1 mg of intramuscular or subcutaneous cyanocobalamin per day (week 1), 1 mg twice weekly (week 2), 1 mg/ week for 4 weeks, and then 1 mg per thirty days for life (about 15%, or one hundred fifty �g, is retained 48 hours after every 1-mg cobalamin injection). Ideally, this protocol for speedy correction of cobalamin deficiency and complete replenishment of cobalamin shops should be used to begin with for all sufferers with cobalamin deficiency, whatever the etiology (see field on Modified Therapeutic Trials). Parenteral hydroxocobalamin must be reserved for all inborn errors of cobalamin metabolism. There is equivalence between oral 2-mg cobalamin tablets consumed daily (where cobalamin is passively absorbed at excessive doses) and traditional month-to-month parenteral treatment with 1 mg of intramuscular/subcutaneous cobalamin among those requiring long-term cobalamin. The reversibility of neurologic harm is slow (a maximal response might take 6 months). Substantial increments (in recovery) are unlikely to be gained after the primary 12 months of acceptable remedy. However, most neurologic abnormalities have improved in up to 90% of patients with documented subacute mixed degeneration. ProphylaxisWithCobalamin Infants on specialised dietsa Premature infants Infants of mothers with pernicious anemiaa Infants and youngsters of moms with nutritional cobalamin deficiency Vegetarianism and poverty-imposed near-vegetarianisma Total gastrectomyb ProphylaxisWithFolicAcidc All women considering pregnancy (at least four hundred �g/day)d Pregnancy and lactation, untimely infants Mothers in danger for supply of infants with neural tube defectse,f Hemolytic anemias/hyperproliferative hematologic states Patients with rheumatoid arthritis or psoriasis on remedy with methotrexateg Patients on antiepileptic drugs Patients with ulcerative colitis For vegetarians, prophylaxis with cobalamin (5- to 10-�g tablet/day) orally ought to suffice. In all other conditions involving any abnormality of cobalamin absorption, cobalamin tablets of 1000 �g/day ought to be administered orally to ensure that cobalamin transport by passive diffusion across the gut is enough to meet daily needs. This method nonetheless serves a function in that enchancment in neurologic standing can be carefully documented. Follow-up outpatient visits each 6 months should be instituted to ensure adequate maintenance of hematopoiesis, as well as early diagnosis of other illnesses generally related to the cobalamin- or folate-deficient state. Patients with pernicious anemia are susceptible to growing iron deficiency that arises from poor iron absorption from achlorhydria. The associated antral enterochromaffin cell hyperplasia (driven by hypergastrinemia that accompanies atrophic physique gastritis) could be related to dysplasia, a risk factor for growth of neoplasia. Supplementation with folate throughout being pregnant additionally helps to forestall premature supply of low-birth-weight infants,15 and routine supplementation for premature infants and lactating moms can be really helpful. Supplementation with folic acid protects in opposition to the event of colorectal neoplasia in high-risk patients with ulcerative colitis. Whereas folic acid supplements consumed in extra of 1 mg/day can mask hematologic signs of cobalamin deficiency, it has been discovered that 94% of U. A robust case can be made to consider the fortification of flour (or different contextually relevant food car in growing countries) with small amounts of cobalamin to serve the majority of the population whose dietary intake of animal-source foods is poor. Immigrants from developing international locations typically proceed to eat their native food plan (which is usually low in animal-source foods) after resettling within the United States; this was earlier documented amongst Indian doctor trainees in New York. Such a diet predisposes to a mixture of dietary cobalamin and folate (and iron) deficiencies, significantly amongst women and youngsters. With cobalamin deficiency, the failure in utilization of intracellular folate for one-carbon metabolism, results in folate leaking out of cells, thereby elevating the serum folate. This is a surprisingly common error in the contemporary literature-a incontrovertible reality that inadvertently downplays (with disastrous consequences) the seriousness of folate deficiency amongst tens of millions of these notably susceptible ladies and children. Once once more, in such settings, the serum folate focus will yield normal-to-high values, predictably underestimate the tissue folate status, and serve to masks the analysis of mild-to-moderate folate deficiency in these patients who desperately require folate to assist compensatory hematopoiesis and for growth and development (See Table 39. The only answer to appropriately figuring out whether these sufferers are in danger for nutritional folate deficiency is to acquire a great dietary history and establish these in danger for nutritional insufficiency. From the sensible standpoint, all people at risk for dietary anemia caused by deficiency of iron, folate, and cobalamin (including each adults150 and children288) must be given prophylactic oral cobalamin, folate (and iron) replacement; and in malarious zones, these must be combined with antimalarial drugs and insecticide-treated mattress nets. There are often additional congenital or acquired causes for hemolytic anemia in a single particular person, similar to malaria, bacteremia, hemoglobinopathy, or glucose-6-phosphate dehydrogenase deficiency.

Buy 20 mg deltasone free shipping
Future efforts ought to be initiated to improve the care of adult patients with thalassemia allergy shots guillain barre syndrome deltasone 40 mg cheap mastercard, together with understanding the pathology of the disease and training new generations of hematologists with an understanding of thalassemia syndromes allergy testing images cheap 40 mg deltasone with visa. Currently, the most common explanation for mortality in adults with thalassemia is related to cardiac illness and arrhythmias. These patients additionally produce other persistent illnesses together with cardiomyopathy, hepatic illness, endocrinopathies, osteoporosis, hypogonadism, pulmonary hypertension, and ongoing infectious risk from splenectomies. Regular follow up with an inner drugs main care doctor can be beneficial to make sure adults are conscious of present screening and treatment pointers relevant to the final inhabitants, for example age-appropriate cancer screening, an space where adult sufferers could not have beforehand been referred because of traditionally shorter lifespans. ExperimentalTherapies Enhancement of and Gene Expression Active -globin genes are hypomethylated in utero but are methylated and inactive after birth. Hypomethylation of the -globin genes could be induced by the drug 5-azacytidine; certainly, short-term administration of this drug produced the anticipated effect in vivo. Preparatory chemotherapeutic regimens to improve immunosuppression and eradicate thalassemic clones utilizing hydroxyurea, azathioprine, fludarabine, busulfan, and cyclophosphamide have elevated the survival fee of sophistication 3 sufferers to 93%; the rejection rate fell to 8%. A section I human gene therapy scientific trial for hemoglobinopathies was initiated in France in 2007. However, the therapeutic Hb in this patient contributed solely one-third of the whole Hb synthesized; the embryonic and fetal Hbs accounted equally for the remaining Hb. Multiple medical trials are at present underway using both gene therapy and genome enhancing technologies as curative approaches for thalassemia main. ThalassemiaIntermedia Approximately 10% of sufferers with homozygous -thalassemia exhibit a phenotype characterized by intermediate hematologic severity. For instance, homozygous -thalassemia in African Americans, Portuguese, and other populations may be relatively delicate, a minimum of for the first twenty years of life. In other words, distinguishing between thalassemia main and thalassemia intermedia, which in turn is the excellence between initiating a regular transfusion program or not, requires consideration of the Hb degree and the standard of life (Table 40. Some nontransfused patients have normal progress and sexual improvement, few medical problems, and normal or near-normal survival rates. However, others develop disfiguring facial changes, markedly delayed growth and sexual maturation, heart failure, extreme osteoporosis, repeated fractures, arthritis, and large splenomegaly. Nonetheless, many households and physicians are reluctant to initiate a chronic transfusion program because of concern about the risks of long-term transfusion therapy and the inevitable want for iron chelation remedy. The want for normal transfusions often develops in adults with thalassemia intermedia because of an additional decline within the Hb level or a rising intolerance of the anemia. By the third or fourth decade, the entire iron burden may attain the levels seen in transfusion-dependent patients. Deferoxamine, 564 PartV RedBloodCells deferiprone, and deferasirox have all proved to be secure and efficient in thalassemia intermedia. A hypercoagulable state can also contribute to the pulmonary hypertension that commonly occurs in sufferers with thalassemia intermedia and is the first cause of congestive coronary heart failure. This complication occurs much less regularly in patients with thalassemia main because of the partial suppression of erythropoiesis by common transfusions. Masses of extramedullary hematopoietic tissue develop in the spinal epidural space, thorax, cranium, pelvis, and elsewhere. For example, sufferers with extramedullary hematopoietic lots may develop paraplegia from spinal wire compression or loss of visible acuity or visual fields brought on by optic nerve compression. The Hb level averages 1 or 2 g/dL lower than that seen in regular persons of the identical age and gender. Hb F ranges decline more slowly than traditional in the first year of life, and the diagnostic elevated Hb A2 levels are established by roughly 6 months of age. Because iron deficiency might happen during pregnancy, iron supplementation has been suggested to keep away from compounding the causes of anemia. Studies have advised there may be an elevated tendency for gallstones and cholecystitis, but in any other case this condition should be largely asymptomatic. However, the gene deletions answerable for the commonest varieties are readily detectable by molecular biology strategies. In basic, partial deletions are extra deleterious and create a more extreme phenotype than full deletions. Nondeletional forms of -thalassemia, which account for 15% to 20% of patients, come up from mutations similar to these described for -thalassemia. The Quong Sze -globin chain (125LeuPro) is exceedingly labile and is destroyed so quickly after its synthesis that no Hb tetramers containing the mutant chain could be shaped. Hemoglobin Constant Spring is an -globin chain variant synthesized in such small quantities (1�2% of normal) that it has the phenotypic impression of a extreme nondeletion -thalassemia allele; nonetheless, the cs allele is at all times linked to a functioning -globin gene, so it has never been related to hydrops fetalis. During the new child period, small quantities (3%) of Hb Bart (4) can be seen by electrophoresis or other strategies. This condition is most often acknowledged when an apparently normal individual becomes the parent of a child with Hb H disease after mating with a person with �-thalassemia trait. The delicate extra of -globin chains might be removed in erythroblasts by proteolysis. At the molecular level, +thalassemia has been discovered to be related to two common gene deletions resulting from different nonhomologous crossing-over occasions between the two linked -globin genes: a 3. Non-deletional HbH occurs when two deletional alpha(0) mutations occur with an alpha (+) mutation. Nondeletional Hb H genotypes usually tend to have larger share Hb H, extra splenomegaly, and more advanced illness. Affected fetuses are usually born prematurely and are either stillborn or die shortly after birth. These morphologic abnormalities and a adverse Coombs test outcome exclude hemolytic illnesses caused by blood group incompatibility. A minor part recognized as Hb Portland (22) migrating within the position of Hb A can additionally be seen. Extreme extramedullary erythropoiesis happens in response to the profound hypoxia and hemolytic anemia attribute of this disease. The common edema attribute of the hydrops fetalis syndrome is a reflection of severe congestive coronary heart failure and hypoalbuminemia in utero. This is partly a consequence of anemia, but the strikingly abnormal oxygen affinity of the tetrameric Hb Bart might be an important determinant of the extreme tissue hypoxia. The oxygen dissociation curve of Hb Bart lacks the traditional sigmoid type because of noncooperativity during oxygen loading and unloading and is markedly shifted to the left. The shift is so great that little oxygen is launched underneath circumstances of low oxygen concentration in the tissues. This Hb does show cooperativity in a manner similar to that of Hb F and due to this fact has a way more favorable oxygen dissociation pattern than that of Hb Bart. A excessive incidence of toxemia of pregnancy has been described in girls carrying severely affected infants, providing an elevated rationale for prenatal prognosis of this condition. Studies of newborns from the archipelago of Vanuatu in the Southwest Pacific and from Papua New Guinea indicate that homozygotes for the rightward -3. Exacerbations of anemia during febrile sicknesses are common and are often characterised by growing fatigue and jaundice. Adults with Hb H develop a few of the same issues as -thalassemia including osteoporosis, cholelithiasis, and iron overload. Hb H may be demonstrated by incubation of blood with supravital oxidizing stains corresponding to 1% sensible cresyl blue.
Woundwort (Goldenrod). Deltasone.
- Spasms; swelling (inflammation) of the mouth, throat, and lower urinary tract; wounds; gout; arthritis; kidney stones; skin conditions; tuberculosis; diabetes; enlargement of the liver; hemorrhoids; internal bleeding; asthma; hayfever; and prostate enlargement.
- Dosing considerations for Goldenrod.
- How does Goldenrod work?
- Are there any interactions with medications?
- Are there safety concerns?
- What is Goldenrod?
Source: http://www.rxlist.com/script/main/art.asp?articlekey=96128

20 mg deltasone cheap with amex
Methemoglobin is fashioned repeatedly allergy forecast jerusalem israel 5 mg deltasone buy fast delivery, but lowering mechanisms keep the methemoglobin level at about 1% of the total hemoglobin allergy report oklahoma order 20 mg deltasone. The consequent left shift of the oxygen dissociation curve will increase hemoglobin affinity for oxygen, thus leading to decreased supply of oxygen into the peripheral tissues and compensatory polycythemia. Epidemiology Most instances of methemoglobinemia are acquired, resulting from increased methemoglobin formation by numerous exogenous agents. Acute or poisonous methemoglobinemia could happen in the setting of overdose or poisoning, but additionally at normal doses of drugs. Acute methemoglobinemia happens equally between men and women and over a extensive range of ages; however, infants are extra vulnerable because their erythrocyte b5R exercise is generally 50% to 60% of grownup exercise. Other causes of hereditary methemoglobinemia are the autosomal dominant inheritance of an abnormal hemoglobin in hemoglobin M disease (see Chapter 43) and, very hardly ever, deficiency of cytochrome b5. Death sometimes ensues at methemoglobin levels above 70% however can occur at lower ranges. Individuals with type I b5R deficiency, which is restricted to erythrocytes, have methemoglobin concentrations of 10% to 35% and seem cyanotic however are usually asymptomatic, even with levels up to 40%. Other neurologic symptoms could also be present, together with microcephaly, opisthotonus, athetoid actions, strabismus, seizures, and spastic quadriparesis. LaboratoryManifestations the laboratory analysis of methemoglobinemia is based on evaluation of its absorption spectra. A recent specimen should all the time be obtained as a end result of methemoglobin ranges are probably to increase with storage. Traditional pulse oximetry is unreliable in the presence of methemoglobinemia because of its light absorbance properties; nevertheless, co-oximetry can decide the methemoglobin fraction along with all other substances with the optical density at 630 nm. Methemoglobin detected by co-oximeter should be confirmed by the particular Evelyn-Malloy method if out there. This technique includes direct spectrophotometric evaluation and should be used when methemoglobinemia is suspected. This methodology remains essentially the most correct approach for the estimation of methemoglobin concentration. Distinguishing the hereditary types of congenital methemoglobinemia requires interpretation of household pedigrees as properly as biochemical analyses. Cyanosis in successive generations suggests autosomal dominant hemoglobin (Hb) M illness, whereas normal parents however possibly affected siblings implies autosomal recessive b5R deficiency. Because the enzyme defect is found in fibroblasts, analysis of b5R exercise in cultured amniotic cells for prenatal diagnosis is feasible. Pathobiology Acute Methemoglobinemia Many medication and toxins have been implicated in acute methemoglobinemia. More widespread culprits include dapsone, native anesthetics (benzocaine, lidocaine, prilocaine), and derivatives of the anesthetic phenacetin. Exposure to nitrates and nitrites, widely used as food preservatives and found in nicely water can also trigger methemoglobinemia. Infants less than 6 months of age might have elevated susceptibility to methemoglobinemia a minimal of in part because of their decrease b5R activity. Homemade child meals purees of high-nitrate-containing greens, nicely water contaminated by nitrites and diarrheal sickness may all trigger acute poisonous methemoglobinemia in infants. B5R Deficiency In erythrocytes, b5R transfers electrons to methemoglobin to reduce it to hemoglobin. In different cells, b5R transfers electrons from cytochrome b5 to stearyl-CoA within the endoplasmic reticulum, a response that has an essential position in ldl cholesterol biosynthesis, fatty acid elongation and desaturation, and drug metabolism. The more common type I b5R deficiency is usually attributable to missense mutations leading to decreased stability of the enzyme. Other pigments, together with methylene blue, may produce false positive outcomes when methemoglobin is measured by co-oximetry. ClinicalManifestations Methemoglobinemia causes clinically discernible cyanosis when absolutely the stage of methemoglobin exceeds 1. Methemoglobinemia ought to be clinically suspected when "cyanosis" happens within the presence of a normal PaO2. Symptoms develop secondary to impaired tissue oxygenation and the onset may be abrupt. At greater levels, respiratory depression, altered consciousness, shock, seizures, and demise could occur. As methemoglobin levels rise above 20% to 30%, Prognosis Acute methemoglobinemia usually resolves promptly with therapy providing the offending trigger is discontinued. Therapy Offending agents in circumstances of acquired methemoglobinemia should be discontinued. However, if the affected person is symptomatic or if methemoglobin ranges are greater than 20%, specific therapy is indicated. Leukomethylene blue, in flip, nonenzymatically reduces methemoglobin to hemoglobin. The cyanosis in hereditary b5R deficiency is of beauty significance solely but may be treated with methylene blue or ascorbic acid, both of which facilitate the reduction of methemoglobin through alternate pathways. Beutler E: Red cell metabolism: a manual of biochemical strategies, ed three, 1984, Grune & Stratton. A abstract of current understanding and management of clinical and metabolic features of pyruvate kinase deficiency. Summary of purple cell enzyme defects in descending order of their clinical significance. This evaluation focuses on the impact of energy metabolism of erythrocyte and its pathophysiology. Viprakasit V, Ekwattanakit S, Riolueang S, et al: Mutations in Kruppellike issue 1 trigger transfusion-dependent hemolytic anemia and persistence of embryonic globin gene expression. Review of pink cell enzymes with in depth bibliography of authentic and up to date articles. In sufferers with severely dysfunctional spectrin mutations, the weakened spectrin dimer-dimer self-association disrupts the skeletal lattice, leading to a marked skeletal instability and cell fragments. It is speculated that elliptocytes are permanently deformed cells as a end result of the weakened horizontal interactions facilitate a shear stress-induced rearrangement of skeletal proteins, precluding restoration of the traditional biconcave shape. Acanthocytosis,Stomatocytosis,andtheBilayer CoupleHypothesis the mechanism of acanthocytosis and stomatocytosis associated with defects of membrane proteins is much less clear. Most forms of acanthocytosis are related to both acquired or inherited abnormalities of membrane lipids. In rare instances with acanthocytosis, membrane protein abnormalities have been detected, however the related mechanisms leading to acanthocyte formation are unknown. These abnormalities happen in the McLeod phenotype, the choreaacanthocytosis syndrome, and different uncommon disorders. In acanthocytosis erythrocytes, brokers that interact with the lipids of the inside lipid bilayer leaflet normalize the form. Vertical interactions, that are perpendicular to the aircraft of the membrane, stabilize the lipid bilayer. These interactions embody spectrinankyrin�band 3 interactions, spectrin-protein 4. Horizontal interactions, which are parallel to the aircraft of the membrane, support the structural integrity of erythrocytes after their publicity to shear stress.
Purchase 10 mg deltasone mastercard
This means that paclitaxel dose escalation in shorter schedules could result in disproportionate increases in space under the concentration�time curve and peak plasma concentration allergy medicine nursing deltasone 5 mg generic on line. Total fecal and urinary excretion of paclitaxel and its metabolites is approximately 70% and 10% allergy shots better than pills purchase deltasone 40 mg with amex, respectively. The drug or its metabolites also have high fecal (80%) and low urinary elimination (5%). Absorption, Fate, and Excretion: Taxanes usually are admin- to 30% of sufferers in the early phase I research. With higher doses of paclitaxel (250 mg/m2 over 24 hours), this could be ameliorated with subsequent administration of granulocyte colony-stimulating issue. Symmetric, distal, peripheral sensory neuropathy is normally seen with higher doses or a number of doses of paclitaxel. Higher doses can also cause motor and autonomic neuropathy as well as myalgias, especially in patients with preexisting neuropathy or when paclitaxel is used with cisplatin. Severe peripheral neuropathy or myalgias are less common after repetitive docetaxel at one hundred mg/m2. Cardiac rhythm abnormalities, particularly bradyarrhythmias and (rarely) coronary heart blocks, have been reported secondary to paclitaxel remedy. A direct causal hyperlink between paclitaxel and myocardial ischemic episodes and tachyarrhythmias has not been established. Although noted, a direct link has additionally not been established between the prevalence of cardiac conductance abnormalities or ischemia and docetaxel remedy. Nausea, vomiting, diarrhea, and stomatitis are uncommon and customarily gentle to reasonable. It is characterised by an erythematous pruritic maculopapular rash affecting the forearms and hands. Onychodystrophy with discoloration, ridging, and brittleness of fingernails also happens. Docetaxel could cause cumulative fluid retention, resulting in peripheral edema, third-space fluid assortment, and weight acquire, which normally resolves slowly after stopping docetaxel. Concurrent treatment with dexamethasone, as famous earlier, delays the onset and decreases the incidence of these unwanted facet effects. Docetaxel for injection is on the market as a concentrate in polysorbate 80 in two vial contents (23. The use of carboplatin after paclitaxel has been reported to trigger less thrombocytopenia than carboplatin alone. Mucositis is more pronounced when paclitaxel is used before doxorubicin, a sequence that reduces the clearance of doxorubicin. Hematologic toxicity is more distinguished with the sequence of cyclophosphamide followed by paclitaxel compared with the reverse sequence of administration. Anticonvulsants such as phenytoin and phenobarbital induce the metabolism of paclitaxel and docetaxel by the P450 mixed-function oxidases. Conversely, in vitro studies have shown that inhibitors of the P450 system can interfere with the metabolism of both medication. These inhibitors embrace erythromycin, testosterone, ketoconazole, and fluconazole. The altered reactivity of the ensuing arabinosyl sugar moiety confers on ara-C its cytotoxic exercise. These embody alopecia, an exfoliative dermatitis, a chemical conjunctivitis (generally ameliorated by the prophylactic administration of a steroid or saline ophthalmic solution), a respiratory distress� like syndrome (characterized by the appearance of rales, irregular radiography findings, and pulmonary insufficiency), and cerebellar toxicity. The latter, which is characterised by nystagmus, ataxia, and different cerebellar signs, could additionally be irreversible, and its appearance mandates discontinuation of remedy. Intrathecal administration of ara-C has been hardly ever related to the toxicities described later for methotrexate. Chemistry and Mechanism of Action: Cytosine arabinoside Potential Drug Interactions: None reported. Approximately 90% of the administered ara-C dose is excreted by the kidneys as ara-U or other inactive metabolites. Methotrexate Chemistry and Mechanism of Action: Methotrexate (N-[4[[(2,4-diamino-6-pteridinyl)methyl]methylamino]benzoyl]- L glutamic acid) represents a member of a category of compounds referred to as antifolates. Methotrexate is a potent inhibitor of dihydrofolate reductase, an enzyme responsible for the reduction of dihydrofolates to tetrahydrofolates. As in the case of most antimetabolites, methotrexate is primarily lively towards S-phase cells. Polyglutamylation of methotrexate enhances its intracellular retention and in some research has been proven to correlate with the sensitivity of leukemic cells to this agent. The deadly actions of methotrexate may be reversed by lowered folates corresponding to 5-formyltetrahydrofolate (leucovorin). The risk that tumor cells may exhibit impaired transport of such decreased folates serves as the idea for methods involving administration of high-dose methotrexate in conjunction with leucovorin rescue. Preparation and Administration: Ara-C is supplied as a sterile, lyophilized powder for reconstitution in vials containing 100 mg, 200 mg, 1 g, or 2 g of material. The powder is reconstituted with sterile bacteriostatic water for injection with benzyl alcohol (0. When reconstituted in this method, options are steady for as a lot as 48 hours beneath managed temperatures. Toxic Effects: Ara-C is primarily toxic to quickly dividing tissues; consequently, myelosuppression and gastrointestinal toxicity characterize the most important unwanted aspect effects of this agent. Patients receiving ara-C often expertise leukopenia, anemia, and thrombocytopenia, with nadirs appearing 7�14 days after drug administration. Gastrointestinal toxicity contains nausea and vomiting, belly pain, mucositis, and a chemical hepatitis characterized by elevation of liver perform enzymes. Methotrexate bioavailability approximates one hundred pc for parenteral routes of administration; with these routes, peak plasma methotrexate ranges are achieved within 30�60 minutes after administration. Methotrexate competes with reduced folates for transport throughout cell membranes; nevertheless, at excessive doses. The terminal half-life of methotrexate is 4�10 hours for patients receiving low-dose remedy and 8�15 hours for these receiving high-dose therapy. Because of the first renal rate of excretion and the potential for nephrotoxicity, methotrexate must be withheld or administered at decreased doses in sufferers with impaired renal operate. Patients receiving high-dose methotrexate therapy ought to be hydrated and their urine alkalinized earlier than administration to cut back the risks of toxicity. For high-dose remedy, leucovorin rescue is required to forestall vital toxicity. If, after 48 hours serum methotrexate levels are higher than 5 � 10-7 M however less than 1 � 10-6 M, leucovorin is continued at a dose of 25 mg/m2 each 6 hours for eight doses till methotrexate levels decline to beneath 5 � 10-7 M.
Generic 20 mg deltasone visa
It is formulated as a sterile allergy attack 5 mg deltasone purchase mastercard, nonpyrogenic allergy forecast toronto buy deltasone 40 mg fast delivery, clear resolution of arsenic trioxide in water for injection using sodium hydroxide and dilute hydrochloric acid to adjust to pH eight. The infusion length may be prolonged up to four hours if acute vasomotor reactions are noticed. The imply elimination half-life of bortezomib after the first dose ranged from 9 to 15 hours at doses starting from 1. The main metabolic pathway is deboronation to kind two deboronated metabolites that subsequently endure hydroxylation to several inactive metabolites. Preparation and Administration: Bortezomib for injection is equipped as a lyophilized powder for reconstitution. Intact vials of lyophilized bortezomib for injection are stored in a fridge at 2�8�C (35�47�F) and protected from gentle. Toxic Effects: probably the most generally reported opposed occasions are asthenic situations (including fatigue, malaise, and weakness; 65%), nausea (64%), diarrhea (51%), decreased appetite (including anorexia; 43%), constipation (43%), thrombocytopenia (43%), peripheral neuropathy (including peripheral sensory neuropathy and peripheral neuropathy aggravated; 37%), pyrexia (36%), vomiting (36%), and anemia (32%). Fourteen percent of sufferers skilled no much less than one episode of grade four toxicity, with the most typical being thrombocytopenia (3%) and neutropenia (3%). Potential Drug Interactions: No formal drug interaction studies have been carried out with bortezomib. In vitro studies with human liver microsomes point out that bortezomib is a substrate of cytochrome P450 3A4, 2D6, 2C19, 2C9, and 1A2. Patients on oral antidiabetic brokers receiving bortezomib treatment could experience hypo- or hyperglycemia and require close monitoring of their blood glucose ranges and adjustment of the dose of their antidiabetic treatment. Finally, patients ought to be cautioned about the use of concomitant medications that may be associated with peripheral neuropathy. Therapeutic Indications in Hematology: Bortezomib is Carfilzomib Chemistry and Mechanism of Action: Carfilzomib is an epoxomicin derivate that irreversibly binds to and inhibits the chymotrypsin-like activity of the 20S proteasome. Inhibition of proteasome-mediated proteolysis results in an accumulation of polyubiquinated proteins, which may lead to cell cycle arrest, induction of apoptosis, and inhibition of tumor progress. It has speedy clearance with an elimination half-life of lower than 30 minutes and a clearance higher than liver blood flow, which suggests there are multiple clearance pathways. Toxic Effects: Carfilzomib has been shown to produce mild-tomoderate nausea and diarrhea. Long-term suppression of gastric acid secretion by H2 blockers or proton pump inhibitors might cut back dasatinib exposure, and antacids are most popular. Preparation and Administration: Ibrutinib is an approved agent available as an oral dose. Absorption, Fate, and Excretion: Bleomycin is rapidly distributed all through the physique and concentrates in the pores and skin, lungs, kidneys, peritoneum, and lymph nodes. Within 24 hours of injection, roughly 50% of an administered dose is excreted unchanged in the urine. Bleomycin elimination correlates well with creatinine clearance; accordingly, patients with renal failure should obtain decreased doses. Preparation and Administration: Dasatinab is an oral agent often taken twice day by day with out regard to meals. Toxic Effects: the most severe poisonous impact is interstitial pneumo- Toxic Effects: Treatment with dasatinib is associated with extreme thrombocytopenia, neutropenia, anemia, and platelet dysfunction. Also seen is fluid retention, including pleural and pericardial effusion, pulmonary edema, extreme ascites, and generalized edema. Pulmonary toxicity is more common in patients older than 70 years, in those receiving a complete dose of larger than 400 U, and in those that acquired prior radiotherapy to the lung. It is essential to emphasize, nonetheless, that the pulmonary toxicity is unpredictable; it has been reported in patients who had none of these danger components and has occurred in a affected person after administration of only 20 U. Some stories recommend that an increased focus of impressed oxygen acts synergistically with bleomycin to produce pulmonary fibrosis. During crucial illness and perioperatively, therefore, an try ought to be made to keep the impressed oxygen concentration at 21%. The early phases of the pulmonary toxicity are clinically manifested by dyspnea and nice rales. Mucocutaneous toxicity occurs in 50% of sufferers treated and is manifested by hyperpigmentation, pruritic erythema, mucositis, desquamation of the plantar surface pores and skin of the arms or toes, ridging of the nails, and alopecia. Febrile reactions, which happen a number of hours after bleomycin administration and will final 4�12 hours, are also frequent. Fever turns into much less frequent with continued use of the drug and may often be prevented by concurrent administration of glucocorticosteroids. Anaphylactoid reactions are noticed in roughly 1% (up to 8% in some series) of sufferers with lymphomas handled with bleomycin. Potential Drug Interaction: Bleomycin, administered with different medication for the treatment of lymphorrhea, can lower the oral bioavailability of digoxin and the pharmacologic impact of phenytoin and certain anesthetic medicine. In 25% or fewer of patients, cerebral dysfunction, characterized by confusion, stupor, and frank coma, can happen. Acute pancreatitis, which can progress to severe hemorrhagic pancreatitis, could occur in 15% of patients. Elevation of liver enzymes and serum bilirubin is almost universal and is histologically represented by fatty metamorphosis. Liver toxicity, although often not clinically vital, has resulted in occasional fatalities. Asparaginase can often produce renal functional impairment with oliguric renal failure. Asparaginase hydrolyzes serum asparagine to nonfunctional aspartic acid and ammonia, depriving tumor cells of a required amino acid; thus, tumor cell proliferation is blocked by the interruption of asparagine-dependent protein synthesis. Chemistry and Mechanism of Action: Asparaginase incorporates instantly before or concurrent with methotrexate, it decreases the cytotoxic effect of the latter. When administered to patients with acute leukemia 9�10 days earlier than or shortly after methotrexate, however, asparaginase appears to enhance the cytotoxic effect of methotrexate. The results of asparaginase on liver operate may probably intervene with the activation or metabolism of other cytotoxic brokers. Therapeutic Indications in Hematology: Asparaginase is used Glucocorticoids Chemistry and Mechanism of Action: Glucocorticoids are artificial compounds derived from the natural adrenal hormone cortisol. Glucocorticoids mediate their biologic actions predominantly by binding to their cytosolic receptor, which then translocates to the nucleus. Lymphocytes treated with glucocorticoids endure apoptosis mediated by glucocorticoid receptors. An early cytostatic part is marked by progress inhibition and cessation of proliferation caused by inhibition of mobile uptake of glucose, amino acids, and nucleosides, as properly as inhibition of macromolecular synthesis. Absorption, Fate, and Excretion: Many synthetic glucocorticoids can be found, the three most commonly utilized in hematology being prednisone, dexamethasone, and methylprednisolone. Glucocorticoids are nicely absorbed orally and are primarily metabolized within the liver. Unlike the opposite two glucocorticoids, the exercise of prednisone depends on hepatic conversion to the 11-hydroxy type (prednisolone). Whereas the biologic half-lives of prednisone and methylprednisolone are roughly 12�36 hours, dexamethasone has a biologic half-life of 36�72 hours.

Deltasone 5 mg order amex
Clinical signs of unstable hemoglobins also rely allergy research group 10 mg deltasone purchase free shipping, partly allergy symptoms pressure deltasone 40 mg proven, on the quantitative proportion of the abnormal hemoglobin. Because the -globin genes are duplicated, mutations in a person locus typically produce only 25% to 35% abnormal globin. By distinction, a easy heterozygote at the single -globin locus usually produces roughly 50% of the irregular variant. The frequent pathway to decreased solubility invariably includes weakening of the binding of heme to globin. Actual loss of heme teams can occur, for instance, in Hb Gun Hill, during which 5 amino acids, including the F8 histidine, are deleted. In different instances, mutations that introduce prolines into helical segments disrupt the helices and intrude with normal folding of the polypeptide across the heme group. Another feature of those mutations is disruption of the integrity of the tetrameric structure of globin chains. Only the intact hemoglobin tetramer can stay dissolved at the excessive concentrations that should be achieved inside the circulating purple blood cell (see Chapters 30 and 40). The basic step in pathogenesis seems to be derangement of the conventional linkages between heme and globin. Loss of acceptable globin chain folding and interaction could in the end destabilize the hemeglobin linkage or result in partial proteolysis of the chain, thereby releasing heme from that linkage. Once freed from its cleft, heme probably binds nonspecifically to other regions of the globin molecule, forming precipitated hemichromes, which outcomes in further denaturation and aggregation of the globin subunits. These form a precipitate containing - and -globin chains, globin fragments, and heme, referred to as the Heinz physique. Heinz bodies work together with delicate pink blood cell membrane elements (see Chapters 43 and 45), thereby lowering purple blood cell deformability. These rigid cells are inclined to be detained within the splenic microcirculation and "pitted," reflecting attempts by the splenic macrophages to take away the Heinz our bodies. Red blood cell harm could be aggravated by the release of free heme into the purple blood cell. Several biochemical perturbations correlate with the presence of free heme, similar to technology of reactive oxidants. AbnormalSolubility 6 GluVal 6 121 121 GluLys GluGln GluLys IncreasedOxygenAffinity ninety two ArgGln J-Capetown 141 89 99 ArgHis SerAsn AspAsn Suresnes Creteil Kempsey DecreasedOxygenAffinity ninety four AspAsn Titusville 102 102 AsnThr AsnSer Kansas Beth Israel Methemoglobin fifty eight HisTyr 87 28 63 67 ninety two HisTyr LeuGln HisTyr ValGlu HisTyr M-Boston, M-Osaka M-Iwate St Louis M-Saskatoon M-Milwaukee-I M-Hyde Park premature destruction of the red blood cell, producing hemolytic anemia. Individual unstable hemoglobins range of their propensity to generate Heinz our bodies and hemolysis. Hemolysis is minimal in nonstressed sufferers with this variant and becomes clinically apparent only in the presence of further oxidant stresses, similar to infection, fever, or the ingestion of oxidant brokers. Nonetheless, the presence of a optimistic family history can be a useful adjunct to analysis and will provoke consideration of an unstable hemoglobin as the purpose for the familial hemolytic diathesis. The scientific syndrome associated with unstable hemoglobin disorders is often referred to as congenital Heinz body hemolytic anemia. This time period derives from the reality that only essentially the most severe instances have been detected earlier than the widespread availability of refined methods Partial record includes a few of the most generally studied hemoglobin structural mutations. Clinical manifestations are highly variable, starting from a just about asymptomatic state within the absence of environmental stressors, to extreme hemolytic anemia manifesting at birth. Most of these that have been described occur on the -chain at invariant residue websites, near critical intermolecular contacts, or in proximity to the prosthetic heme-binding web site. For hemoglobin variants with a given degree of decreased solubility, the diploma of anemia might fluctuate because some of these variants also exhibit altered oxygen affinity. Thus Hb K�ln has elevated oxygen affinity, resulting in comparatively higher ranges of tissue hypoxia and erythropoietin stimulation at any given stage of hematocrit (see Diagnosis); due to this fact sufferers with Hb K�ln tend to have greater hematocrit levels than anticipated on the idea of hemolytic severity due to elevated erythropoietin stimulation. By contrast, Hb Hammersmith displays decreased oxygen affinity, enhancing oxygen supply and allowing sufferers to perform at a decrease hematocrit degree. Hb Zurich possesses, for complicated molecular causes, a higherthan-normal affinity for carbon monoxide. A high hemoglobin carbon monoxide level develops in patients with Hb Zurich who also smoke. Binding of carbon monoxide protects Hb Zurich from denaturation, thus lowering hemolysis, so these people are probably to exhibit lesser degrees of hemolytic anemia than do nonsmoking relations. Diagnosis the presence of an unstable hemoglobin ought to be suspected in sufferers with one or more stigmata of accelerated red blood cell destruction: persistent or intermittent hemolytic anemia or jaundice, premature development of bilirubin gallstones or biliary tract disease (as a result of accelerated purple blood cell turnover), unexplained reticulocytosis, or bouts of intermittent symptoms that may be associated to exposure to oxidant medicine or infections. Laboratory analysis is dependent upon identification of a mutant hemoglobin that precipitates extra easily than regular hemoglobin. Two provocative laboratory maneuvers are used to help detection, each of which unmask the tendency of unstable hemoglobins to precipitate: the warmth instability test (heating of a hemoglobin solution to 50�C) or the isopropanol instability test (insolubility in 17% isopropanol). For instance, Hb K�ln, the commonest of the unstable hemoglobin mutations, arises from a mutation changing the valine at position ninety eight to a methionine. A regular electrophoretogram, however, should never be regarded as robust proof towards the presence of a mutant hemoglobin, especially if the clinical picture or family historical past otherwise helps the prognosis. Mass spectrometry analysis of hemoglobin and direct globin gene sequencing are supplanting electrophoresis as diagnostic strategies. Additional subtle analyses of hemoglobin may be obtained from reference laboratories if detailed characterization appears warranted. For example, irregular hemoglobin or globin bands migrating to novel positions on an isoelectric focusing gel may finish up from hemoglobin or globin moieties lacking heme in groups. When heme is added to the sample and the proteins are reanalyzed, these bands disappear. Detection of unstable hemoglobins is often compromised by the selective precipitation of the unstable variant into Heinz bodies. Because most sufferers are heterozygotes, this phenomenon tremendously reduces the apparent percentage of the variant in soluble form. Thus even a variant possessing altered electrophoretic mobility may be very difficult to detect. Indeed, some unstable hemoglobins, such as Hb Geneva or Hb Terre Haute, are so unstable that no mutant gene product could be detected within the steady state. They are detectable only by isotope labeling studies or direct evaluation of the globin genes. The amino acid sequence predicted from genetic sequencing could not often be inaccurate because of posttranslational conversion into an unstable hemoglobin. Through posttranslational modification, the methionine is altered to aspartate, a hydrophilic residue that disrupts the heme pocket. The differential analysis of unstable hemoglobin variants is often straightforward if this common category of hemolytic problems is suspected. This analysis ought to be thought-about, as ought to other causes of chronic or intermittent hemolytic anemia, including red blood cell membrane issues.
Buy cheap deltasone 20 mg on line
Even in sufferers without silent or overt cerebral infarction food allergy treatment 2013 10 mg deltasone generic, cognitive functioning can be impaired allergy treatment vivite vibrance therapy by allergan deltasone 10 mg buy overnight delivery. Less well-documented however potentially modifiable risk factors embrace alcohol or drug use, oral contraceptive use, and sleep-disordered breathing. Intracranial hemorrhage leads to the same signs as thrombosis, however in addition, neck stiffness, photophobia, severe headache, vomiting, and altered consciousness could happen. Although the mortality rate could additionally be as excessive as 50%, the morbidity of survivors is low. Hemorrhage may be subarachnoid, intraparenchymal, or intraventricular, which could be differentiated by angiography. The favorable neurosurgical outcome in subarachnoid hemorrhage caused by ruptured aneurysm justifies an aggressive approach to diagnosis, transfusion, vasodilatory therapy, and surgery. Over a period of more than 2 years, the danger of stroke was decreased to lower than 1% per year within the transfused group171 (a risk reduction of >90%). The capability of transfusion to curtail development of large-vessel stenosis has additionally been proven with angiography. This trial evaluated discontinuation of transfusion after no much less than 30 months in kids who had not had an overt stroke and in whom the cerebral move rates decreased to low danger (<170 cm/s) with transfusion. Other modifiable threat components for stroke (see Cerebrovascular Accidents, Pathophysiology, Incidence, Risk Factors, and Presentation) must be identified and handled. Notably, in the common population, hypertension is particularly related to a threat for hemorrhagic stroke, and efficient remedy of hypertension can produce a relative risk reduction of 26% for ischemic stroke and 49% for hemorrhagic stroke. Patients with systolic pressures within the higher range for the sickle cell group, even with systolic pressures less than a hundred and forty mmHg, had an elevated danger of first ischemic stroke (there were insufficient events to make firm conclusions concerning hemorrhagic stroke). This remedy additionally offers incidental safety towards ache crises, bacterial infections, acute chest syndrome, and hospitalization. This is most likely not possible for administrative causes or because of allosensitization or iron overload for which the affected person is unable or unwilling to endure remedy. In patients with moyamoya illness, surgical approaches to therapy, such as extracranial�intracranial bypass, have been useful in enhancing the perfusion of affected areas of the mind. Stem cell transplantation has resulted in stabilization of cerebral vasculopathy,178 however the threat of a second neurologic event is greater within the peritransplant interval, and the mortality price with this process is between 6% and 10%. In each thrombosis and hemorrhage, prompt partial-exchange transfusion is carried out, and continual direct transfusion to maintain the Hb S stage below 30% is instituted to prevent recurrent events (see additionally Basic Management and Disease Modification) and promote decision of arterial stenoses. In one examine, 21 of 152 sufferers in a pediatric clinic had seizures, 4 of which were related to meperidine therapy. The widespread acute issues are pneumonia and acute chest syndrome, and the frequent persistent complication is pulmonary hypertension. Antibiotic remedy for pneumonia or acute chest syndrome ought to cover these brokers in addition to pneumococcus and H. However, it ought to be borne in mind that the identical old etiology could be each vasoocclusion and infection simultaneously, and in virtually all instances of acute chest syndrome, antibiotics ought to be administered. Pulmonary fats embolus, evidenced by stainable fats in pulmonary macrophages obtained by bronchoalveolar lavage or sputum induction, is found in 44% to 60% of circumstances of acute chest syndrome. Some patients have a rapidly progressive course related to a precipitous decrease in arterial oxygen rigidity; they might require intensive care therapy. Chronic issues similar to pulmonary hypertension happen in as many as 60% of patients. Blood gasoline and pulmonary perform measurements must be obtained as baseline data for all sufferers. The degree rises after the primary decade, probably because of continual hepatobiliary dysfunction. Alkaline phosphatase ranges are elevated in all genotypes till puberty, which occurred later in males and in those with sickle cell anemia. Some have really helpful the surgical elimination of asymptomatic gallstones to keep away from subsequent issue in distinguishing gallbladder ache from acute painful episodes. This approach has turn out to be extra feasible with the provision of laparoscopic cholecystectomy. This syndrome normally resolves within 3�14 days with supportive care alone but can progress to liver failure and fatal consequence, due to this fact patients must be monitored intently and trade transfusion initiated in the occasion that they present indicators of progressive liver dysfunction. Acute Hepatic Sequestration Crisis Acute hepatic sequestration disaster presents with acute hepatic enlargement and a dramatic fall in Hb concentration, the most probably mechanism being sequestration of sickled erythrocytes within the liver. Intrahepatic Cholestasis Maternal problems embrace increased charges of painful episodes, severe anemia attributable to iron or folate deficiencies, exaggeration of the physiologic "anemia of being pregnant," elevated infections (urinary tract infections, pneumonias, endometritis), preeclampsia, and death. The incidence of a perinatal death in a previous pregnancy and the presence of twins within the current pregnancy are two main threat factors for an unfavorable consequence. Better fetal and maternal outcomes in current times are largely attributable to typically improved antenatal and obstetric care. Patients should be followed in a high-risk obstetric clinic in addition to the hematology clinic and obtain the standard vitamin, mineral, and folate dietary supplements. Some consultants recommend prophylactic transfusion, but a big controlled study showed no enchancment in fetal end result from this administration possibility, although maternal signs are lowered. If the Hb is between 8 and 10 g/dL and transfusion is indicated for any of the explanations above, partial exchange must be performed. Some consultants advise that hypertonic saline injections are contraindicated for elective termination of pregnancy due to the chance of sickling-induced vasoocclusion. There are anecdotal stories of a higher incidence of acute painful episodes after therapeutic abortion; inpatient intravenous hydration before and for the 24 hours after the procedure is really helpful. Sickle cell intrahepatic cholestasis leads to severe, asymptomatic hyperbilirubinemia without fever, ache, leukocytosis, hepatic failure, or demise. Evidence of progressive liver dysfunction ought to prompt consideration of acute hepatic cell disaster and change transfusion. Liver transplantation has been used efficiently as therapy for this complication. Pregnancy entails elevated dangers to the mother and baby compared with the general inhabitants. Another warning with low-dose estrogen oral contraception is the risk of contraceptive failure with lower than excellent compliance. There could also be dangers to contraception, however towards this must be weighed the dangers of unintended pregnancy. Sexually energetic girls ought to have routine pelvic examinations and birth control directions. Renal Complications Hypertension, proteinuria, hematuria, rising anemia, and nephrotic syndrome reliably predict development to renal failure, that are scientific indices to pay consideration to as a outcome of the serum creatinine may be misleading. Patients with sickle cell anemia exhibit an elevated proximal tubular secretion of creatinine. These sufferers usually survive and recuperate their renal operate with no increased threat of creating chronic renal failure.